Abstract
Dermatitis herpetiformis (DH) is a cutaneous manifestation of gluten sensitivity. Although over 90% of DH patients have evidence of a gluten-sensitive enteropathy, only about 20% have intestinal symptoms of celiac disease (CD). Both the skin disease and the intestinal disease respond to gluten restriction and recur with institution of a gluten-containing diet.
Four findings support the diagnosis of DH:
- •
pruritic papulovesicles or excoriated papules on extensor surfaces
- •
neutrophilic infiltration of the dermal papillae with vesicle formation at the dermal–epidermal junction
- •
granular deposition of IgA within the dermal papillae of clinically normal-appearing skin adjacent to a lesion
- •
a response of the skin disease, but not the intestinal disease, to dapsone therapy.
Although DH is usually a lifelong condition, the course may wax and wane. A spontaneous remission may occur in up to 10% of patients, but most clinical remissions are related to dietary gluten restriction.
Linear IgA bullous dermatosis (LABD) is an immune-mediated, subepidermal, vesiculobullous eruption that occurs in both adults and children. It has been defined on the basis of a unique immunopathology consisting of linear deposition of IgA along the cutaneous BMZ. In adults, the clinical findings associated with this immunopathologic pattern may resemble those of DH or bullous pemphigoid (BP), but in children, the cutaneous features may be clinically unique.
Keywords
dermatitis herpetiformis, dapsone, linear IgA bullous dermatosis, LABD, DH, gluten, celiac disease, gluten-sensitive enteropathy, granular IgA deposition, linear IgA deposition, Linear IgA dermatosis, chronic bullous disease of childhood
- ▪
Dermatitis herpetiformis (DH) is differentiated from linear IgA bullous dermatosis (LABD) on the basis of granular IgA versus linear IgA deposition at the basement membrane zone, as observed by direct immunofluorescence
- ▪
DH is a cutaneous manifestation of celiac disease (CD) and is associated with gluten sensitivity in virtually all cases
- ▪
DH and CD are genetic disorders strongly associated with the HLA-DQ2 genotype, in which IgA anti-endomysial antibodies are directed against tissue transglutaminase; the presumed autoantigen within the skin is epidermal transglutaminase
- ▪
The intestinal and cutaneous disease in DH can be controlled by gluten restriction; both DH and LABD respond to sulfone therapy
- ▪
In adults, LABD is often drug-induced
Dermatitis Herpetiformis
▪ Duhring disease
Introduction
Dermatitis herpetiformis (DH) is a cutaneous manifestation of gluten sensitivity. Although over 90% of DH patients have evidence of a gluten-sensitive enteropathy, only about 20% have intestinal symptoms of celiac disease (CD). Both the skin disease and the intestinal disease respond to gluten restriction and recur with institution of a gluten-containing diet.
Four findings support the diagnosis of DH:
- •
pruritic papulovesicles or excoriated papules on extensor surfaces
- •
neutrophilic infiltration of the dermal papillae with vesicle formation at the dermal–epidermal junction
- •
granular deposition of IgA within the dermal papillae of clinically normal-appearing skin adjacent to a lesion
- •
a response of the skin disease, but not the intestinal disease, to dapsone therapy.
History
Dr Louis Duhring described DH at the University of Pennsylvania in 1884. It was the first skin disease described by an American dermatologist. The subsequent critical discoveries in our understanding of DH are listed in Table 31.1 .
| DERMATITIS HERPETIFORMIS – HISTORIC LANDMARKS | |
|---|---|
| 1884 | Louis Adolphus Duhring of Philadelphia described in detail “dermatitis herpetiformis” (DH) |
| 1888 | Jean Louis Brocq of Paris renamed the condition “dermatite polymorphe prurigineuse” |
| 1890 | T Caspar Gilchrist outlined the histologic changes of DH, which were included in the 1897 edition of Duhring’s textbook Cutaneous Medicine: A Systemic Treatise on Diseases of the Skin |
| 1940 | Sulfapyridine was reported to be an effective treatment for DH. The clinical response to sulfapyridine became a diagnostic test |
| 1950 | Dicke, a Dutch pediatrician, observed that patients with celiac disease (CD) improved during World War II when bread was in short supply, and their disease worsened when grain supplies were restored |
| 1953 | Diamino-diphenyl sulfone (dapsone), the parent compound of sulfapyridine, also proved to be effective in DH |
| 1969 | JB van der Meer demonstrated IgA deposits in the papillae of uninvolved skin in DH |
| 1967 | Independent accounts connected DH with CD |
| 1972 | Stephen Katz linked DH to the HLA-B8 antigen. This strengthened the association between CD and DH |
| 1986 | IgA class anti-endomysial antibodies discovered to be highly specific for DH and CD |
| 1997 | DH and CD found to have a common immunogenetic background with strict association with HLA alleles DQ A1*0501 and B1*02, which encode HLA-DQ2 heterodimers |
| 2003 | Epidermal transglutaminase was identified as an autoantigen in DH |
| 2012 | Zone and colleagues demonstrated that transfer of DH sera or goat anti-human epidermal transglutaminase antibodies to human skin-grafted mice mimics DH immunopathology |
Epidemiology
DH occurs most commonly in people of northern European origin. It is uncommon in African-Americans and Asians. According to a study in Finland in 1978, the prevalence of DH is 10.4 per 100 000 individuals and the annual incidence is 1.3 per 100 000. The mean age of onset is during the fourth decade, but it may first appear from age 2 to 90 years. Adolescent and prepubescent children are infrequently affected. In most epidemiologic studies, DH occurs more frequently in men as compared to women, in a ratio of 1.1 : 1 to 1.9 : 1 . From 1979 to 1996, the familial incidence of DH in Finland was studied prospectively. DH was diagnosed in 1018 patients and 10.5% of these patients had one or more affected first-degree relatives .
With regard to the US, a study in Utah (1987) found a prevalence of 11.2 per 100 000. The annual incidence during the years 1978 to 1987 was 0.98 per 100 000. Mean age of onset for men was 40.1 years and for women was 36.2 years. The male-to-female ratio was 1.44 : 1 .
Pathogenesis
Our knowledge of the pathogenesis of DH is based on a number of clinical and laboratory observations. The key observations that have been integrated into theories of pathogenesis are:
- •
a strong genetic association with the HLA genotype DQ A1*0501, B1*02 (which encodes HLA-DQ2 heterodimers), in addition to other unidentified non-HLA genes
- •
some degree of gluten-sensitive enteropathy on small bowel biopsy in virtually all patients, accompanied by stimulation of the mucosal immune system
- •
granular IgA deposition within the papillary dermis of the skin (this is essential for the diagnosis and occurs at the site of eventual inflammation)
- •
neutrophilic infiltration into the dermal papillae
- •
improvement of symptoms with dapsone therapy and worsening of symptoms with inorganic iodide ingestion.
Genetic predisposition
Specific HLA genes, which encode molecules that interact with T-cell receptors, are believed to provide the antigenic specificity that processes the gliadin antigen in genetically susceptible individuals. This HLA association is the same for patients with CD and DH. Genes encoding the DQ2 (A1*0501, B1*02) heterodimer are carried by 90% of CD and DH patients, while genes encoding the DQ8 (A1*03, B1*03) heterodimer are carried by the remaining DH patients . Previous descriptions of associations with HLA-B8, as well as HLA-DR3 and HLA-DR5/DR7, represent class I and class II molecules that are in linkage dysequilibrium with DQ2. However, it has been established that <50% of the genetic predisposition in CD and DH is due to specific HLA genes. Genomic searches for non-HLA genes are currently underway and a susceptibility locus for CD (chromosome 4q27) has been proposed which includes the genes that encode IL-2 and IL-21 . Monozygotic twins, one with DH and the other with CD, have been reported, indicating that environmental as well as genetic factors play a role in the development of CD and/or its extraintestinal manifestation, DH .
Gluten-sensitive enteropathy
On small bowel biopsy, more than 90% of DH patients have some degree of gluten-sensitive enteropathy. The bowel abnormality is caused by gluten, a family of grain proteins present in wheat, rye, barley, and hybrids of these grains (e.g. kamut, spelt, triticale), but not oats. Gliadin represents the alcohol-soluble fraction of gluten and is believed to be the antigenic component. The spectrum of intestinal involvement ranges from minimal infiltration of the lamina propria by lymphocytes (with normal villi), to minimal atrophy of the jejunum accompanied by intraepithelial lymphocytic infiltrates, to total villous atrophy of the small intestine. The enteropathy is often patchy and may require multiple small bowel samples for diagnosis. Symptomatic malabsorption occurs in 20% of patients with DH.
The proposed pathogenesis of DH and CD is presented in Fig. 31.1 . Following ingestion of gluten-containing grains (see above), one of the products of digestion is gliadin ( Fig. 31.1A ). Once gliadin is absorbed via the lamina propria, glutamine residues within gliadin are deamidated by tissue transglutaminase (TG2) and covalent cross-links (isopeptidyl bonds) are formed between lysine residues in TG2 and glutamines in gliadin. Deamidation by TG2 is thought to be a critical step as it serves to optimize antigen presentation.
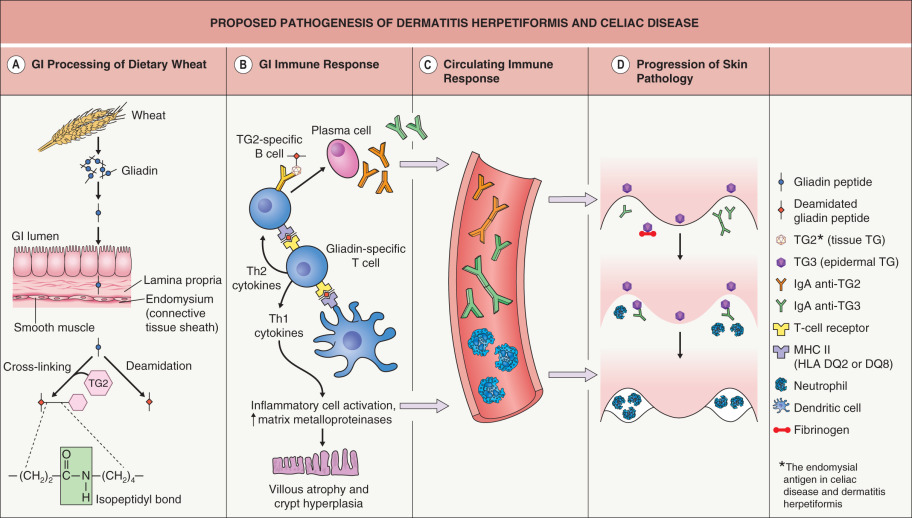
Deamidated gliadin peptides bind to the groove of the HLA-DQ2 molecule on dendritic antigen-presenting cells ( Fig. 31.1B ), and the gliadin antigen is then presented to sensitized helper T cells in the context of HLA-DQ2 specificity. These helper T cells can stimulate B cells, with differentiated plasma cells producing IgA antibodies to multiple antigens, including gliadin, gliadin cross-linked to TG2, TG2, and epidermal transglutaminase (TG3). In addition, stimulated natural killer lymphocytes cause crypt hyperplasia and villous atrophy. Of note, IgA anti-TG2 antibodies have become the serologic hallmark for CD.
In the setting of continued exposure to gliadin, epitope spreading is thought to lead to development of IgA anti-TG3 antibodies in patients who already have IgA anti-TG2 antibodies; a subgroup of those who develop IgA anti-TG3 antibodies then develop DH (see Fig. 31.1 ). Epitope spreading is a possible explanation for why DH tends to present at a later age than symptomatic CD (the latter often manifests in childhood) and why patients with CD (but not DH) tend to have more severe intestinal disease than patients with DH. Formation of IgA anti-TG3 antibodies is thought to require time and continued exposure to gluten and this would be more likely to occur in patients with less severe, relatively asymptomatic intestinal involvement. Findings in support of this theory include the presence of IgA anti-TG2 antibodies in most DH patients and a higher prevalence of IgA anti-TG3 antibodies in adults than in children with CD (i.e. developing later in the evolution of the disease).
The formation of IgA anti-TG3 antibodies also activates circulating neutrophils ( Fig. 31.1C ). Deposition of these antibodies within the dermal papillae results in the infiltration of activated neutrophils from the circulation into the dermal papillae ( Fig. 31.1D ). Degranulation of neutrophils releases proteases which disrupt the lamina lucida and produce a subepidermal blister.
Since both the skin disease and the intestinal disease resolve with dietary gluten restriction and recur with return to a regular diet, it is clear that the dietary protein gluten is central to the pathogenesis of the cutaneous eruption. In addition, it is the HLA class II antigen that acts as a gate through which gluten can reach the inflammatory cells and initiate the autoimmune process .
Circulating antibodies
The first serologic difference between DH and CD was initially described in 2002, with TG3 identified as the autoantigen in DH . Additional studies have demonstrated that circulating IgA anti-TG3 antibodies are not only elevated in patients with DH, but can be assayed and may be helpful in monitoring response to a gluten-free diet . TG3 was also found to co-localize with IgA in dermal papillae of DH patients and is enzymatically active in this site (see Fig. 31.5B ) . TG3 is expressed in many tissues of the body, including the epidermis. When IgA anti-TG3 antibodies reach the dermis, they complex with TG3 antigens which have been produced by keratinocytes (epidermal TG) and then have diffused into the dermis. In other words, IgA/TG3 immune complexes are formed locally within the papillary dermis (see Fig. 31.1D ).
When goat anti-human TG3 antibodies (IgG) were passively transferred into SCID mice grafted with human skin, immune deposits were detected within the papillary dermis which reacted with rabbit anti-TG3 and DH sera. Transfer of sera from DH patients (who had high circulating levels of IgA anti-TG3 antibodies) led to similar granular IgA and TG3 dermal deposits . In DH skin, IgA-bound deposits of TG3 are enzymatically active and therefore the TG3 likely plays an important role in the covalent binding of IgA to connective tissue fibers. Enzymatically active TG3 also binds to soluble fibrinogen whose subsequent degradation may play a key pathogenic role .
Granular IgA deposition
Granular IgA deposition within dermal papillae is the hallmark of DH (see Fig. 31.5A ). The deposits are composed of IgA1 antibodies directed against TG3 antigen that has diffused from the epidermis. Circulating IgA antibodies to TG2 (an endomysial antigen) have been identified by indirect immunofluorescence microscopy using a monkey esophagus substrate, and the presence of these antibodies correlates with the degree of gluten-sensitive enteropathy. IgA anti-TG2 antibodies are not responsible for the IgA deposition in skin.
Iodide and dapsone
Ingestion of iodide can lead to worsening of DH and topical application of iodide onto the normal skin of DH patients produces lesions that are histologically identical to spontaneous lesions . Even in normal subjects, topical iodides may produce follicular neutrophilic pustules. The mechanism by which iodide stimulates neutrophil infiltration into the skin is unclear.
Dapsone is known to have an effect on neutrophil chemotaxis and neutrophil attachment to IgA in vitro . Although the exact mechanism of its beneficial effect in DH is unknown, it seems likely that dapsone blocks the neutrophil-mediated inflammatory process.
Associated Disorders and Malignancies
A strong association exists between DH and thyroid disease, particularly Hashimoto thyroiditis. In one study, the presence of some type of thyroid abnormality was found in 26 of 50 patients with DH . Although the pathogenetic relationship between DH and thyroid disease is unknown, it may represent a coexistence of autoimmune disease . The incidence of enteropathy-associated T-cell lymphoma is also increased in patients with DH and warrants increased surveillance. Of note, adhering to a gluten-free diet protects against lymphoma in this population. This further supports advising patients with DH to adhere to a strict gluten-free diet for life. Other autoimmune disorders reported to be associated with DH and CD are listed in Table 31.2 .
| AUTOIMMUNE DISORDERS ASSOCIATED WITH DERMATITIS HERPETIFORMIS | |
|---|---|
| Common | |
| |
| Uncommon | |
| |
| Rare | |
|
|
Clinical Features
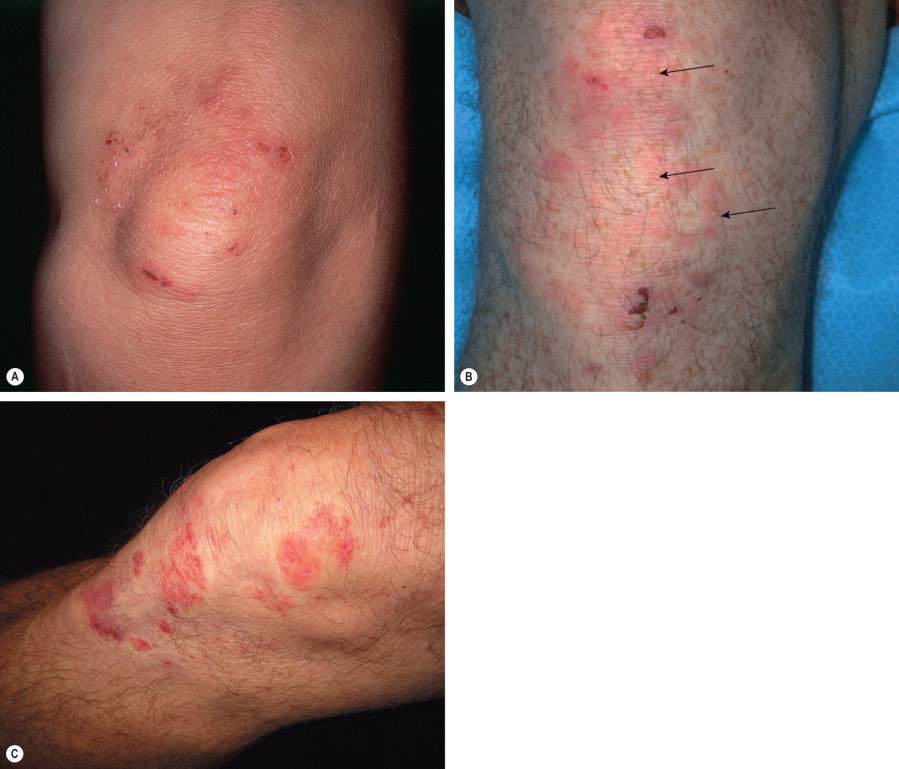
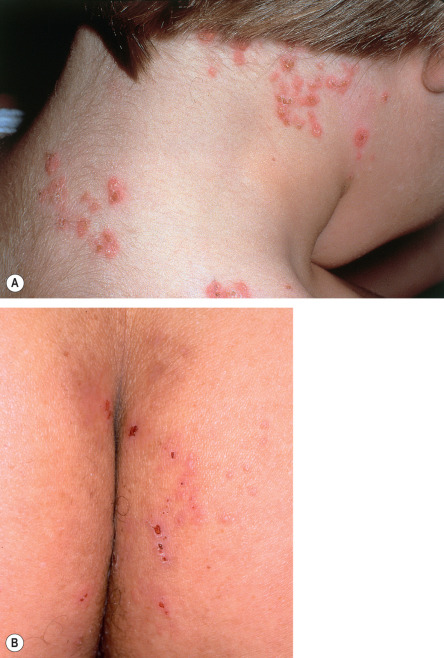
DH has a symmetric distribution and favors the elbows, knees, extensor forearms, back, and buttocks ( Figs 31.2 & 31.3 ). Isolated scalp involvement is an occasional clinical presentation. The primary lesions are pleomorphic, with urticarial plaques, papules, and vesicles. Grouped or “herpetiform” papulovesicles with an erythematous base are characteristic of this autoimmune disorder. Intense pruritus leads to secondary excoriation and crusting. Even if only hemorrhagic crusts or secondary changes from scratching are present, the diagnosis should be suspected on the basis of the distribution pattern. Less common presentations are isolated facial involvement, exclusively macular lesions, and hemorrhagic acral macules (usually palmoplantar).
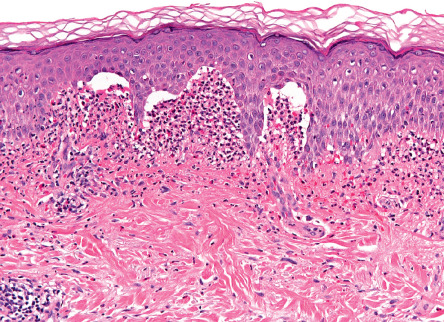
Pathology
For routine histology, it is optimal to capture a small, intact vesicle. If this is not available, an area of erythema should be biopsied. Areas of erythema will show dermal papillary edema and neutrophil infiltration associated with a superficial perivascular lymphocytic infiltrate. Dermal papillae filled with neutrophils, with relative sparing of the lowermost tips of the intervening rete ridges, is a characteristic finding. When an intact vesicle is biopsied, a subepidermal blister containing predominantly neutrophils will be seen ( Fig. 31.4 ).
In patients with DH, dermal IgA deposits are not uniformly distributed throughout the skin. Notably, IgA deposits are present in greater amounts near active lesions. The optimal biopsy site for direct immunofluorescence (DIF) testing is normal-appearing skin immediately adjacent to a lesion. A false-negative DIF can occur if lesional skin is biopsied, since the inflammatory infiltrate can destroy the IgA . A definitive diagnosis of DH cannot be made without this diagnostic DIF finding. Granular IgA deposits localized to the dermal papillae are found in 85% of cases of DH ( Fig. 31.5 ), while continuous granular deposition of IgA along the basement membrane occurs in 5% to 10% of cases. A fibrillar pattern of IgA deposition occurs rarely.
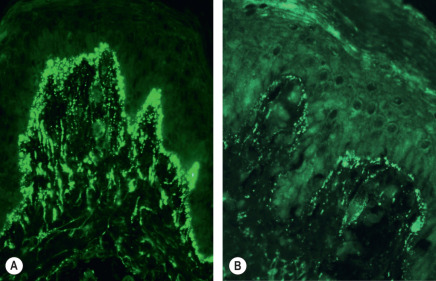
The lack of histologic and immunopathologic confirmation is a source of misdiagnosis and confusion in DH. Since DH is a lifelong diagnosis with significant systemic and therapeutic implications, definitive immunopathologic confirmation of the diagnosis is essential in all cases.
Laboratory evaluation
Anti-endomysial antibodies are very specific for CD and DH. Their levels indicate the severity of the gluten-sensitive enteropathy and reflect the degree of compliance to dietary gluten restriction. Anti-endomysial antibodies are found in ~80% of patients with DH and >95% of patients with active CD. The endomysium is the connective tissue covering the smooth muscle layers of the esophagus, stomach, and small intestine (see Fig. 31.1A ). Studies have confirmed a high sensitivity and specificity for IgA class anti-reticulin and anti-endomysial antibodies in DH patients with villous atrophy.
The endomysial antigen has been identified as TG2, and antibodies directed against transglutaminase are important in the pathogenesis of DH and CD (see Pathogenesis section). The normal physiologic function of transglutaminase is to repair injured tissue by cross-linking extracellular matrix proteins in the tissue, thus protecting the surrounding tissue from further damage. Transglutaminase facilitates the linkage of the carboxamide group of the amino acid glutamine, found in gluten, to an ε-amino group of a lysine residue in transglutaminase located in the intestinal tract (see Fig. 31.1A ). The glutamine in gliadin is especially vulnerable to a cross-linkage with transglutaminase, and this linkage is perceived as a foreign antigen by the host immune system. As antibodies to transglutaminase are produced, the normal function of transglutaminase in repairing the damaged intestinal mucosa is impaired.
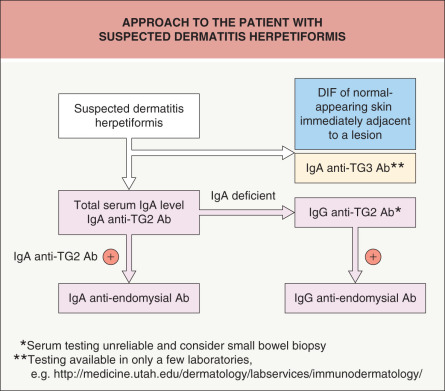
An approach to the patient with suspected DH is outlined in Fig. 31.6 .