Fig. 9.1
Varying clinical appearances of congenital melanocytic nevi. (a) A medium-sized, evenly pigmented congenital melanocytic nevus on the arm. (b) A large heterogeneous congenital melanocytic nevus on the scalp and upper back. (c) A giant posterior axial congenital melanocytic nevus
CMN are classified according to the maximum diameter the nevus is expected to obtain in adulthood: small <1.5 cm; medium ≥1.5–19.9 cm; and large ≥20 cm. In newborns, the projected adult size can be obtained by multiplying the diameter by the following factor determined by location: 1.7 (head); 2.8 (torso or upper extremities); or 3.5 (lower extremity).
Patients with CMN are at a higher risk of malignant melanoma and neurocutaneous melanosis. The exact lifetime incidence of melanoma in CMN remains to be determined, but is estimated to be <1 % in small and medium-sized nevi, and 2–5 % for large nevi [1–3]. The risk of melanoma in small- and medium-sized CMN increases after puberty, and superficial spreading melanoma is the most common presentation. In contrast, half of melanomas occurring in large CMN appear before the age of 5 and are dermal or subcutaneous in origin; these patients are also at risk for extracutaneous melanoma. The risk of melanoma is greatest in patients with giant nevi (>40 cm) in a posterior axial location.
Neurocutaneous melanosis (NCM) refers to the abnormal deposition of nevomelanocytes along the leptomeninges of the brain or spinal cord. Three percent to 10 % of patients with high-risk large CMN develop symptomatic NCM [4]. Symptoms of NCM include seizures, hydrocephalus, developmental delay, and focal neurologic deficits. Symptomatic NCM most commonly presents by the age of 3 and is associated with a poor prognosis. Risk factors for NCM are: (1) >40 cm CMN in a posterior axial location; (2) >20 satellite nevi; and (3) patients with ≥3 medium-sized CMN [5].
The diagnosis of CMN is usually straightforward, but a biopsy can be performed if necessary. Histological examination reveals nests of nevus cells at the epidermal-dermal junction and dermis, often with extension into the deeper dermis and subcutis, around adnexal structures, and single file between collagen bundles. Dermoscopy can be a useful diagnostic tool and typically shows reticular, globular, reticuloglobular, or homogeneous patterns.
Management Strategies
The approach to the management of CMN is individualized and dependent on many factors; including the age of the patient, size and location of the CMN, ease of monitoring for malignant changes, risk of melanoma and NCM, and psychological impact. Surveillance is a reasonable option, given the low absolute risk of melanoma, especially when surgical excision is not feasible due to unacceptable cosmetic or functional outcomes.
Small- and medium-sized CMN can be observed or excised, depending on the above factors. Parents and patients should be counseled on periodic examination and bring to attention any abrupt focal changes in size, color, border, new growths, or new onset symptoms, such as pain or pruritus. Any suspicious changes should be biopsied.
Patients with large CMN require closer monitoring. More frequent examinations, typically every 3–12 months, are necessary for patients with large or giant CMN. Examination should include visual inspection along with palpation for firm, subcutaneous nodules. Any suspicious changes should be evaluated with biopsy. Referral to a surgeon experienced in the removal of CMN should be offered to the family. Excision is thought to decrease, although not eliminate, the risk of melanoma. Regardless of excision, patients need lifelong follow-up and evaluation of scars and any residual nevi.
All patients with CMN should be counseled on the importance of UV protection and self-examination. Some patients may benefit from psychological counseling or support groups.
Investigations Recommended
A biopsy can be done to confirm the diagnosis or to exclude malignant transformation. Baseline and serial photographs and dermoscopy are useful noninvasive tools for monitoring CMN. A MRI of the brain and spinal column should be done in all patients with a large CMN or ≥3 medium-sized CMN, and neurological symptoms to evaluate for NCM, CNS melanoma or other CNS malformations.
Additionally, baseline MRI can be considered in asymptomatic individuals with LCMN >40 cm or >20 satellite nevi or ≥3 medium-sized CMN, although this is debatable. Ideally, MRI screening is best done between 4 and 8 months of age. If the patient has neurological symptoms or the MRI is abnormal, referral to a neurologist is warranted. See Table 9.1 for specific investigations recommended.
Table 9.1
Specific investigations recommended for CMN
For diagnosis |
Biopsy, confirmation of diagnosis or exclusion of malignancy |
Baseline photographs |
Baseline dermatoscopic evaluation |
MRI of brain and/or spine |
In patients with large CMN and neurological symptoms |
Midline lumbosacral location to rule out tethered spinal cord |
Consider baseline in asymptomatic patients with high-risk lesions: giant CMN (>40 cm, posterior axial location) or >20 satellite nevi or ≥3 medium CMN |
Neurodevelopmental assessment |
Referral to neurologist, if indicated |
Pet scan, if MRI suspicious for deep or extracutaneous melanoma |
For surveillance |
Periodic examination by physician (large CMN every 3–12 months, small or medium CMN more important after puberty) |
Serial photographs and dermoscopy |
Monthly self-examination by family and/or patient |
First Line Therapies
Observation is appropriate for the majority of lesions. If treatment is desired for aesthetic reasons, symptoms, or malignancy concerns, full thickness excision is the treatment of choice. For most lesions it is recommended that excision be carried down to the fascia. For larger lesions this may require serial excisions, skin grafts, or the use of tissue expanders. See Table 9.2.
Table 9.2
First line therapies for CMN
Therapy | Evidence level |
|---|---|
Observation | B |
Full thickness excision | D |
Second Line Therapies
Laser therapy may be an option to improve cosmesis in lesions not amenable to surgical excision. Various lasers, both pigment specific and nonselective, have been used with widely variable efficacy, ranging from poor to excellent. Scarring and dyspigmentation are potential complications and recurrence is very common. Multiple treatment sessions are necessary. There is some concern that laser treatment of CMN may mask the development or even increase the risk of melanoma, but this has not been proven in studies [6–8].
Curettage and dermabrasion have been advocated as procedures to remove the superficial portion of CMN, thereby lightening the lesion and decreasing the nevus cell burden. Results have been variable. Both of these procedures are done in the newborn period to take advantage of the natural cleavage plane between the epidermis and dermis. Recurrence and hypertrophic scarring are potential complications. There is some concern that the scarring induced by these procedures may mask the detection of melanoma (Table 9.3).
Table 9.3
Second line therapies for CMN
Therapy | Evidence level |
|---|---|
Laser therapy | |
Nonselective ablative lasers: CO2 laser and erbium:YAG | D |
Pigment selective lasers: Q-switched ruby, Nd:YAG and alexandrite | B |
Dermabrasion | D |
Curettage | D |
Spitz Nevus
Spitz nevi, or Spitz tumors, typically present in children and adolescents as pink or red well-circumscribed, dome-shaped papules on the face or lower extremities (Fig. 9.2). In adults, Spitz nevi more commonly manifest as brown or black papules. Spitz tumors are most often solitary, though groups of lesions may occur. These lesions may grow rapidly for several months and then stabilize. Composed of large epithelioid and/or spindled cells, they can be difficult to distinguish histologically from melanoma [9–12].
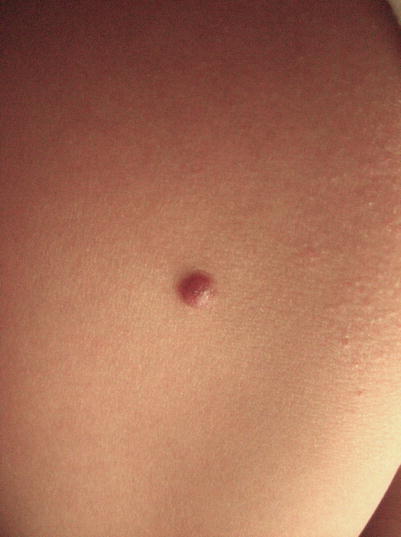
Fig. 9.2
Spitz nevi
Spitz tumors most often develop before 20 years of age and occur in all ethnic groups, with slightly higher predominance among young females. Benign Spitz tumors are usually seen in children younger than 10. Spitz tumors that develop in patients older than 20 years of age carry increased malignancy risk [10–14].
Spitz tumors are classified along a histological and clinical continuum, from conventional benign Spitz tumors to malignant Spitz neoplasms, with pleomorphic features impossible to differentiate from melanoma. Between these extremes exists a range of atypical Spitz tumors, with varying degrees of cytologic atypia and clinical behavior [11].
Conventional Spitz tumors are generally less than 6 mm in diameter, symmetric, and sharply defined. Atypical Spitz tumors are usually 6–10 mm in diameter and may show asymmetry, color variegation, and irregular topography. It is rarely possible to clinically distinguish atypical Spitz tumors from melanoma, as both may contain atypical findings such as large diameter (greater than 1 cm), asymmetric border, and color variegation. This distinction is sometimes made retrospectively after adverse events such as metastasis or death [11–16]. Cases of multiple Spitz nevi have been reported, but they have not been associated with any increased risk of malignancy [17, 18].
Management Strategies
Spitz tumors are diagnosed based on clinical and/or histologic features. Ancillary immunohistochemical and genetic studies exist though without the sensitivity or specificity necessary to distinguish atypical Spitz tumors from melanoma.
Lesions without atypical features should be monitored every 3–12 months. Patients should return if the lesion changes appearance. Lesions concerning for atypical Spitz tumors or melanoma should be removed by simple excision with margins of 3–5 mm (Table 9.4). Atypical Spitz tumors with positive margins should be re-excised with clear margins of 1 cm if cosmetically feasible. Severely atypical Spitz tumors should be managed as melanomas. Sentinel lymph node biopsy in the case of atypical Spitz tumors is controversial, as there is no data to suggest this improves prognosis (Table 9.5).
Specific Investigations
For diagnosis |
Dermoscopic exam |
Skin biopsy for histologic evaluation |
Molecular and cytogenetic testing if histologically indicated |
For treatment |
Monitor every 3–12 months for lesions without atypical features |
Simple excision of atypical Spitz tumors (3–5 mm margins) |
Re-excision of atypical Spitz tumors with + margins (1 cm if possible) |
Dermoscopic Features: Conventional Spitz tumors show little to no pigmentation with a dotted vascular pattern. A “starbust” or “peripheral globular pattern” may occur in pigmented lesions [16].
Histological Features: Conventional Spitz tumors show maturation, a uniform cell population, and rare or no mitoses. Atypical Spitz tumors may show asymmetry, significant cytologic atypia, and dermal mitoses. There are no firm histologic features that distinguish atypical Spitz tumors and melanoma, though melanomas often show greater cytologic pleomorphism and higher mitotic rate [11, 19, 20].
Table 9.4
First line therapies
Observation. Monitor every 3–12 months for lesions without atypical features | E |
Simple excision of atypical Spitz tumors with 3–5 mm margins especially in those over 12 years of age | C [21] |
A retrospective review of 89 Spitz tumors found that biopsy or excision with less than 3 mm margins was associated with positive margins in greater than 20 % of cases [21].
*A retrospective analysis of ten recurrent Spitz tumors using comparative genome hybridization found that two cases yielded results consistent with melanoma, rather than Spitz nevi [11].
Halo Nevus
Clinical Features
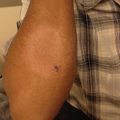
A halo nevus is a melanocytic nevus surrounded by a zone of depigmentation, often resulting in spontaneous regression of the nevus [22]. This halo phenomenon is most often associated with acquired benign melanocytic nevi, but has been reported to occur around congenital nevi (Fig. 9.3), Spitz nevi, blue nevi, and melanoma. Halo nevi can be single or multiple, and most frequently occur on the trunk. They are common in children and young adults, with an estimated prevalence of 1 %; but occur at higher rates in patients with vitiligo and Turner syndrome [23, 24].
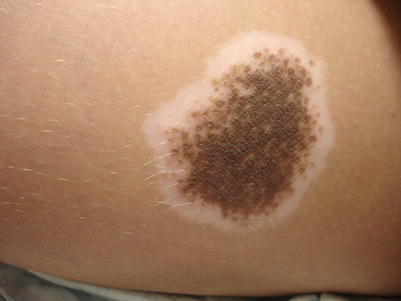
Fig. 9.3
A depigmented halo surrounding a medium-sized congenital melanocytic nevus
The diagnosis is usually evident clinically. Dermatoscopic examination most commonly shows a globular and/or homogeneous pattern encircled by a white structureless area [25]. Histological examination shows a dense, often band-like, infiltrate of lymphocytes in the papillary dermis intermingled with nests of nevus cells.
Investigations Recommended
The central nevus should be carefully evaluated using the “ABCD” diagnostic criteria for melanoma. Dermatoscopic examination is also recommended. A full skin examination is useful for evaluating all of the patient’s nevi. A skin biopsy of the central nevus is indicated if there are worrisome clinical and/or dermatoscopic features. See Table 9.6 for a summary of recommended investigations.
Table 9.6
Specific investigations recommended for Halo Nevi
For diagnosis |
Close clinical evaluation of the central nevus and halo |
Dermatoscopic examination |
Full skin examination |
Biopsy, if the central nevus or halo has atypical clinical or dermatoscopic features |
First Line Therapies
Observation is generally sufficient for halo nevi. Most are benign and spontaneously regress over time. If the central nevus or surrounding halo has any concerning features, a biopsy is indicated and further treatment is based on the histological findings. See Table 9.7.
Table 9.7
First line therapies for Halo Nevi
Therapy | Evidence level |
|---|---|
Observation | D |
Biopsy, if central nevus or halo atypical | D |
Becker’s Melanosis
Clinical Features
Becker’s melanosis (also known as Becker’s nevus) is a cutaneous hamartoma which often develops in the peripubertal period, although has been noted in some cases to be present at birth [27]. Clinically, this benign hamartoma is typically hyperpigmented and unilateral with an irregular border and surface (Fig. 9.4). Common locations are the shoulders, scapula or anterior chest. In many cases increased overlying hair and/or acne are present. The hypertrichosis and acne tend to be more prominent in males because of androgen mediated effects [28]. Becker’s nevus syndrome has been described which is characterized by associated abnormalities including breast hypoplasia, limb asymmetry, scoliosis, and supernumerary nipples [29].
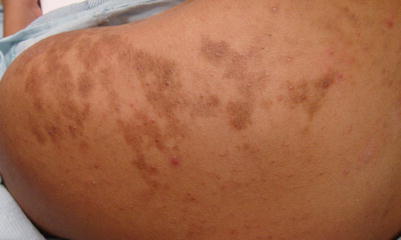
Fig. 9.4
Becker’s melanosis containing acne inflammatory papules
Investigations Recommended
For diagnosis |
Biopsy: The histopathologic findings are acanthosis, papillomatosis and increased pigmentation of the basal layer |
Thorough history and physical to assess for signs of Becker’s nevus syndrome |
Table 9.8
First line therapies
Observation |
Lightening hair |
Lightening pigmentation |
Treat acne |
Becker’s melanosis is a benign hamartoma often identified in the peripubertal period. Identification of the lesion should trigger consideration of Becker’s nevus syndrome. The lesion typically requires no therapy unless it becomes symptomatic.
Tumor of the Epidermal Appendages: Nevus Comedonicus
Clinical Features
Nevus comedonicus is a hamartoma involving the pilosebaceous unit. It consists of grouped dilated follicular openings containing central keratotic plugs, resembling open comedones. Pustules, cysts, abscesses, secondary bacterial infection and scarring can occur. Lesions can be localized or extensive.
Nevus comedonicus syndrome refers to a neurocutaneous disorder that consists of nevus comedonicus with associated skeletal (scoliosis, fused vertebrae, spina bifida), ocular (cataracts), limb defects (absent fifth finger, polydactyly), or neurological abnormalities [31].
A variety of benign epithelial tumors, including trichoepithelioma, syringocystadenoma papilliferum, hidradenoma papilliferum, and keratoacanthoma, have been reported to occur in nevus comedonicus. Malignant tumors are rare, but basal cell and squamous cell carcinoma have been reported [32].
The diagnosis is usually evident clinically. Histological examination shows hairless dilated follicular ostia filled with lamellar keratin, epidermal invaginations, rudimentary sebaceous glands, and absent arrector pili muscles.
Management Strategies
Nevus comedonicus is a benign lesion, therefore treatment is not required, unless desired for aesthetic purposes or for lesions complicated by recurrent inflammation and/or infection. The only definitive treatment is surgical excision. Numerous medical and surgical treatments have been reported, with incomplete responses. The treatment of extensive lesions is very challenging and often disappointing.
Investigations Recommended
A skin biopsy can be done if the diagnosis is uncertain. Patients with extensive lesions should be evaluated carefully for the existence of nevus comedonicus syndrome. See Table 9.10 for specific investigations.
Table 9.10
Specific investigations recommended for nevus comedonicus
For diagnosis |
Biopsy, if diagnosis uncertain |
Exclude nevus comedonicus syndrome in patients with extensive lesions |
Physical examination, with focus on skeletal, ocular, digital and nervous system |
Ophthalmologic evaluation, if indicated |
Roentgenogram of spine, if indicated |
Neurology referral, if indicated |
First Line Therapies
Complete surgical excision is curative and is the treatment of choice for lesions that are bothersome and amenable to surgical excision. Various topical therapies, including emollients, topical keratolytics (ammonium lactate 12 %, salicyclic acid), topical retinoids (tretinoin, tazarotene), topical vitamin D analogs (calcipotriene, talcalcitol), alone or in combination may improve cosmesis in some patients [31–33]. Commercial pore remover strips may be helpful for removing keratin plugs [34]. Inflammatory lesions may respond to treatment with topical or intralesional corticosteroids. Oral antibiotics are indicated if secondary infection is present. Incision and drainage or excision of localized abscesses or cysts is effective (See Table 9.11) [35].
Table 9.11
First line therapies for nevus comedonicus
Therapy | Evidence level |
|---|---|
Localized lesions | |
Surgical excision, curative and best treatment if lesion amenable to surgery | E |
Topical keratolytics (ammonium lactate 12 %, salicylic acid) | E |
Topical retinoids (tretinoin, tazarotene) | E |
Topical vitamin D analogs (calcipotriene, talcalcitol), alone or in combination with topical retinoids | E |
Commercial pore remover strips | E |
Extensive lesions | |
Topical keratolytics, retinoids, vitamin D analogs | E |
Inflamed lesions (pustules, cysts, abscesses) | |
Topical or intralesional corticosteroids | E |
Oral antibiotics | E |
Incision and drainage or excision of localized abscesses or cysts | E |
Second Line Therapies
Laser treatment with various lasers, including 1,450 nm diode laser, ultrapulsed CO2 laser, and erbium:YAG laser, have been reported to be effective in adults. Laser therapy can be considered for bothersome lesions not amenable to surgical excision or for extensive lesions [36, 37].
Isotretinoin has been reported to be helpful for decreasing the number of cystic flares associated with inflammatory lesions, but has not been effective as a therapy for non-inflamed lesions (Table 9.12) [34].
Table 9.12
Second line treatment for nevus comedonicus
Therapy | Evidence level |
|---|---|
Localized or extensive lesions | |
Laser therapy (1,450 nm diode laser, ultrapulsed CO2 laser, erbium:YAG) | E |
Inflammatory lesions | |
Isotretinoin (1 mg/kg), may decrease cystic lesions, otherwise ineffective | E |
Third Line Therapies
Dermabrasion, shave excision, and manual comedone extraction have poor efficacy and often produce unacceptable scarring and recurrence (Table 9.13).
Table 9.13
Third line treatment for nevus comedonicus
Therapy | Evidence level |
|---|---|
Dermabrasion | E |
Shave excision | E |
Manual comedone extraction | E |
Nevus Sebaceous
Clinical Features
Nevus sebaceous (NS) is a hamartoma of the pilosebaceous unit. NS occurs in approximately 0.3 % of newborns and is usually evident at birth as an oval or linear, hairless, yellow-brown plaque on the scalp (Fig. 9.5a) or less frequently on the face and neck. At puberty, NS becomes more papillomatous or verrucous, due in part to androgen stimulation of the aberrant sebaceous glands (Fig. 9.5b).
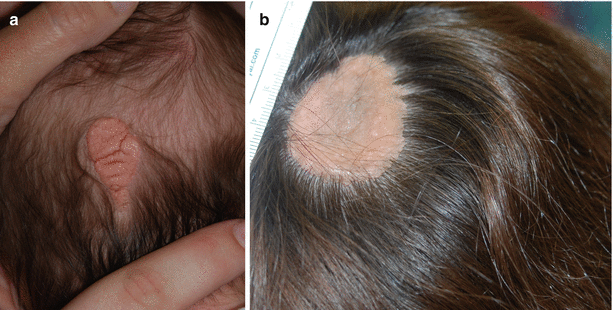
Fig. 9.5
Nevus sebaceous. (a) A nevus sebaceous on the scalp at birth. (b) A nevus sebaceous on the scalp in an adolescent that has become more verrucous
It is estimated that approximately 20 % of lesions will develop secondary, mostly benign, tumors. The most common tumor is trichoblastoma, followed by syringocystadenoma papilliferum. Basal cell carcinoma is estimated to occur in approximately 1 % of lesions and is extremely unlikely to appear before puberty. Squamous cell carcinoma, adenocarcinoma, and sebaceous carcinoma have also been reported [38–40].
Nevus sebaceous syndrome (Schimmelpenning syndrome) refers to the association of an extensive linear nevus sebaceous that follows the lines of Blaschko with neurological (seizures, developmental delay, hemimegalencephaly), ocular (lipodermoid, coloboma), and skeletal (skull deformity, scoliosis, short stature) anomalies. Patients with nevus sebaceous syndrome are also at risk for Vitamin D-resistant hypophosphatemic rickets, which should be suspected in patients with an extensive nevus sebaceous along with growth retardation, bone pain, fractures or bone deformities [41].
The diagnosis of nevus sebaceous can usually be made clinically. A biopsy can be done if the diagnosis is uncertain. In prepubertal children, histopathology reveals mild epidermal acanthosis and papillomatosis with immature sebaceous glands and pilosebaceous units. After puberty, there is more prominent acanthosis and papillomatosis of the epidermis, prominent sebaceous glands often located high in the dermis, immature hair follicles and ectopic apocrine glands in the lower dermis.
Management Strategies
Historically, excision prior to puberty was recommended due to the perceived high rate of basal cell carcinoma. Given that the rate of malignant transformation is actually quite low, especially in children, prophylactic excision of all lesions before puberty is no longer recommended. However, lesions should be observed for suspicious changes, and new localized growths or ulceration should be biopsied and completely excised if necessary. Prophylactic removal may be desirable in some cases for cosmetic concerns, and the timing of such excision should be evaluated on an individual basis.
Investigations Recommended
A skin biopsy is indicated if the diagnosis is uncertain or to exclude malignancy. Patients with an extensive nevus sebaceous should be evaluated carefully for the occurrence of nevus sebaceous syndrome. See Table 9.14 for specific investigations recommended.
Table 9.14
Specific investigations recommended for nevus sebaceous
For diagnosis |
Biopsy, to confirm diagnosis or exclude a secondary malignant neoplasm |
Exclude nevus sebaceous syndrome in patients with extensive linear NS |
Ophthalmologic examination |
MRI of brain, as indicated in patients with suspected neurologic abnormalities |
Skeletal radiographs, if indicated |
Serum and urine calcium and phosphorous levels, parathyroid hormone levels, vitamin D levels, in patients with suspected hypophosphatemic rickets |
First Line Therapies
Full thickness excision, when feasible, with 2–3 mm margins, is the treatment of choice for lesions that are symptomatic or cosmetically concerning. Staged excision or tissue expansion can be done if necessary. The timing of excision is dependent on the age of the child, size and location of the lesion, and the risks and benefits of general anesthesia versus local anesthesia later in life (see Table 9.15) [40].
Table 9.15
First line therapy for nevus sebaceous
Therapy | Evidence level |
|---|---|
Full thickness excision with 2–3 mm margins | D |
Second Line Therapies
Nonsurgical options for the treatment of NS include photodynamic therapy and various lasers. These treatments produce variable results and should be reserved for extensive lesions or lesions not amenable to surgical excision. Photodynamic therapy using various photosensitizers (aminolevulinic acid 20 % or methylaminolevulinate) and light sources (light emitting diodes device, 630-nm argon tunable dye laser, intense pulsed light), have resulted in cosmetic improvement in a small number of patients with facial NS. Laser ablation or curettage of thicker lesions or nodules was done prior to photodynamic therapy in some patients [42, 43]. Different lasers, including continuous wave CO2 laser, fractional CO2 laser, and erbium:YAG laser, have been reported to be useful in single case reports [40, 44]. Multiple treatment sessions were necessary for all of these modalities (Table 9.16). It is important to realize that these modalities do not completely remove the lesion, leaving a risk for recurrence and development of secondary neoplasms; therefore continued monitoring is necessary.
Table 9.16
Second line therapies for nevus sebaceous
Therapy | Evidence level |
|---|---|
Photodynamic therapy | D |
Laser therapy (CO2 laser, fractional CO2 laser, erbium:YAG laser) | E |
Other Adnexal Tumors
Clinical Features
Adnexal tumors are a diverse group of benign neoplasms originating from adnexal epithelium. The most common adnexal tumors in children include pilomatricomas, trichoepitheliomas, and syringomas [45]. These tumors are often solitary, but can be multiple, especially when associated with an underlying syndrome (Table 9.17).
Table 9.17
Syndromes associated with multiple adnexal tumors
Adnexal tumor | Clinical appearance | Associated syndrome |
|---|---|---|
Trichoepithelioma | Firm, flesh-colored papules in central face | Brooke-Spiegler syndrome |
Multiple familial trichoepitheliomas | ||
Cylindroma | Pink, firm plaques or nodules most common on the scalp | Brooke-Spiegler syndrome |
Familial cylindromatosis | ||
Trichilemmoma | Small flesh colored, sometimes verrucous papules, in central face | PTEN hamartoma—tumor syndrome (includes Cowden and Bannayan-Riley-Ruvalcaba syndromes) |
Pilomatricoma | Firm, subcutaneous nodule with pink or blue hue; “teeter-totter” or “tent” sign | Myotonic dystrophy, Gardner syndrome, Rubenstein-Taybi syndrome |
Syringoma | Small flesh-colored to yellow translucent papules | Down syndrome |
Sebaceous adenoma | Yellow, lobulated papules on face | Muir-Torre syndrome |
Pilomatricomas are most common on the head and extremities and present as hard, mobile subcutaneous nodules, often with a pink or blue hue. Due to calcification in the tumor, they may show a “teeter-totter” sign or “tent” sign when the overlying skin is stretched. Trichoepitheliomas appear as skin-colored papules with a predilection for the central face. Syringomas present as small, yellow-white translucent papules most commonly on the lower eyelids, neck, upper chest and genitalia (Fig. 9.6). They may occur in an eruptive manner. The clinical presentation of other adnexal tumors is summarized in Table 9.18.
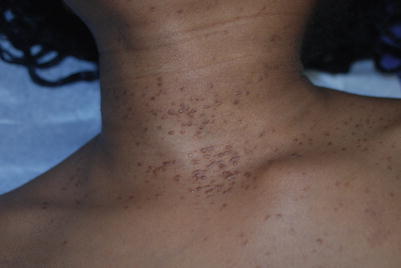
Fig. 9.6
Multiple eruptive syringomas on the neck and upper chest in an adolescent
Table 9.18
Specific investigations recommended adnexal tumors
Skin biopsy, if needed for diagnosis |
Evaluation for potential syndrome in patients with multiple adnexal tumors |
Management
Adnexal tumors are benign and therefore treatment is not necessary, but may be desired for aesthetic or symptomatic reasons. Solitary lesions are best treated by surgical excision.
Treatment of multiple adnexal tumors is often difficult, but electrodessication, electrosurgery, cryosurgery, dermabrasion, chemical peels and laser ablation have been reported with variable success.
Investigations Recommended
A skin biopsy may be necessary for definitive diagnosis. If multiple tumors are present, or there is a positive family history of similar lesions, further work-up to rule out an underlying syndrome may be necessary. See Table 9.18 for specific investigations recommended.
Table 9.19
First line therapies for adnexal tumors
Therapy | Evidence level |
|---|---|
Surgical excision | D |
First Line Therapies
Second Line Therapies
Laser ablation using various CO2 lasers (continuous wave, pulsed, fractional) or erbium:YAG has been reported for patients with multiple syringomas or trichoepitheliomas (see Table 9.20). Several treatment sessions may be necessary, and recurrence and scarring are potential complications [47]. Other destructive methods have also been reported in small numbers of patients.
Table 9.20
Second line therapies for adnexal tumors
Therapy | Evidence level |
|---|---|
Laser ablation (CO2, fractionated laser, erbium:YAG) | B |
Electrodessication | E |
Electrosurgery | E |
Cryosurgery | E |
Trichloroacetic acid, various strengths | E |
Tumor of the Epidermis: Mastocytosis
Clinical Features
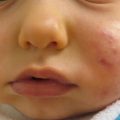
Mastocytosis is characterized by infiltration of mast cells in the dermis. Maculopapular cutaneous mastocytosis lesions (MPCM) have a “peau d’orange” clinical appearance (Fig. 9.7). The lesions urticate and become reddened with rubbing (positive Darier’s sign) and some may become vesicular. Cutaneous lesions of mastocytosis may present as: (1) a single lesion (Solitary mastocytoma); (2) multiple maculopapular lesions (maculopapular cutaneous mastocytosis-MPCM aka Urticaria pigmentosa); or (3) diffuse infiltrative lesions [48].
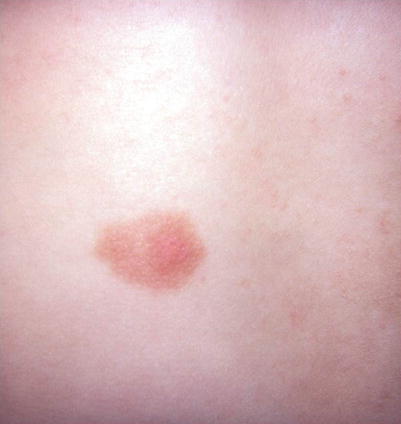
Fig. 9.7
Solitary mastocytoma with “peau d’orange” appearance
The lesions are often present before 2 years of age and may affect any area of the body. Spontaneous resolution of hyperpigmentaiton is often seen by 10–12 years of age, although urticarial changes are usually difficult to elicit after 4 years of age [49].
Familial mastocytosis are rare, with fewer than 100 cases reported in the literature [50]. Individual lesions are clinically similar between different subtypes of cutaneous mastocytosis, however, familiar mastocytosis often persist into adulthood. Over 90 % of patients with mastocytosis have mutations of the c–kit gene that often occur spontaneously. Mutations have been found on exons 8, 9, 10, 11, 13, 17, and 18 of c–kit.
Childhood-onset mastocytosis has a better prognosis than adult-onset disease and has less systemic involvement that is mastocytosis related [48].
Management Strategies
Initiate measures to minimize blistering or discomfort.
Avoid triggers. Some triggers include: vigorous rubbing, hot baths, aspirin, alcohol, ibuprofen, and codeine. A very complete list of potential triggers can be found at the website Mastokids website-www.mastokids.org [51].
Investigations Recommended
For diagnosis |
Biopsy—shows mast cell infiltration in the dermis and around the blood vessels. Giemsa stain and toluidine blue can stain the granules in the mast cells |
Genetic test for c–kit mutations |
Table 9.21
First line therapies
Solitary Mastocytoma | ||
Avoid triggers | ||
Topical corticosteroids | Evidence level E | [52] |
Excision | Evidence level E | [53] |
MPCM | ||
Avoid triggers | ||
Cyproheptadine, H1 blockers | Evidence level B [54] | |
Table 9.22
Second line therapies
H2 blockers | Evidence level C | [54] |
Pimecrolimus cream and antihistamine | Evidence level E | [54] |
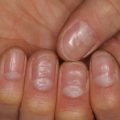
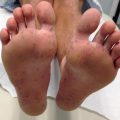
Basal Cell Nevus Syndrome
Clinical Features
Basal cell nevus syndrome (BCNS; also known as Gorlin syndrome) is an autosomal dominant multi-system disorder caused by mutations in the PTCH1 gene. The main features are: (1) multiple basal cell carcinomas (BCCs); (2) odontogenic keratocysts of the jaw; (3) palmar/plantar pits; (4) skeletal abnormalities (most commonly of the ribs and spine); (5) ectopic calcification of the falx cerebri; (6) ocular abnormalities; and (7) medulloblastoma. Macrocephaly and typical facial features, including frontal bossing and hypertelorism, are frequent and early findings. Young children often have multiple facial milia and skin lesions that resemble nevi (“basal cell nevi”) or skin tags, but with the typical histological appearance of BCC (Fig. 9.8) [56]. These lesions generally lack aggressive behavior in childhood. Invasive BCC usually presents during late teens and early adulthood and becomes more frequent with age. Crusting, ulceration, and enlargement are signs of invasion. The diagnosis of a BCC before the age of 20 should signal an evaluation for BCNS.
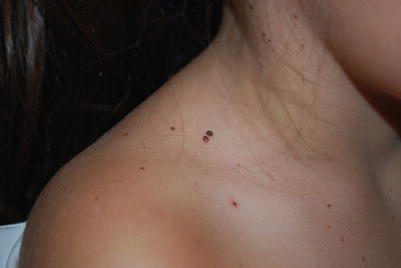
Fig. 9.8
Multiple basal cell carcinomas resembling nevi in a young child with basal cell nevus syndrome
Management
The management of BCNS requires a multidisciplinary and individualized approach, which entails appropriate surveillance for complications, minimizing exposure to radiation, treatment of tumors, and genetic counseling. Several surgical and nonsurgical options exist for the treatment of BCCs. Often a combination of treatment modalities is necessary and the choice of treatment is dependent on many factors, including the age of the patient, location, size and number of BCCs, as well as the histological subtype. Aggressive treatment of BCCs in younger children may not be necessary, as most of these lesions will not become invasive. The optimal treatment of BCCs in patients with BCNS remains to be defined.
Exposure to radiation should be minimized as much as possible as soon as the diagnosis is suspected. Approximately 5–10 % of children develop medulloblastoma and treatment with radiation therapy should be avoided, if feasible, because of the high numbers of BCCs that may develop in the field of radiation. All patients should be counseled on the importance of strict sun protection. This involves the use of protective clothing and glasses, SPF 30+ sunscreens, and avoidance of sun during midday hours. Radiographs should be minimized as much as possible and performed only as needed for diagnosis or management of valid medical issues.
Investigations Recommended
The diagnosis of BCNS is made based on diagnostic criteria. A thorough clinical examination and radiographs of the jaw, skull, chest, and spine are usually necessary to confirm the diagnosis. Modalities utilizing non-ionizing radiation, such as MRI, ultrasound or digital technology, are preferred [57]. Molecular genetic testing is commercially available and is useful for diagnostic confirmation in patients with suspected BCNS not fulfilling diagnostic criteria or in at-risk asymptomatic family members (i.e. child of a parent with BCNS). Skin biopsy is indicated if invasive BCC is suspected. See Table 9.24 for specific investigations recommended for diagnosis and surveillance.
Table 9.24
Specific investigations recommended for basal cell nevus syndrome
For diagnosis |
AP and lateral radiographs of the skull, chest, and spine |
Orthopantogram (by 8 years of age) |
Ophthalmologic examination, baseline |
Echocardiogram (in first year of life)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|