Fig. 24.1
A baby with failure to thrive and poor wound healing after delayed separation of umbilical stump
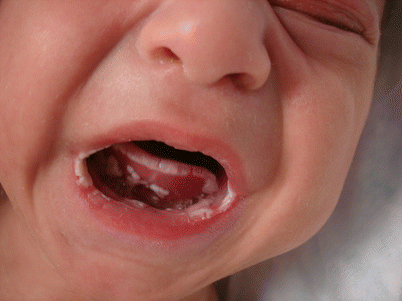
Fig. 24.2
Treatment-resistant thrush in undifferentiated immunodeficiency disorder
Table 24.1
Clinical cutaneous features of primary immunodeficiency disorders
Disorder | Cutaneous features |
|---|---|
Immunoglobulin deficiencies | |
IgA deficiency | Autoimmune-like disorders: vitiligo, atopic dermatitis, lupus, recurrent candida infections, purpura, necrotizing vasculitis, lipodystrophy, and visceral granulomas |
Combined variable immunodeficiency | Verruca, pyoderma, extensive dermatophyte infections, autoimmune diseases as seen in IgA deficiency, sarcoidal and deep granuloma annulare-like granulomas |
X-linked hypogammaglobulinemia | Cutaneous abscesses, furuncles, cellulitis, pyoderma gangrenosum, and dermatomyositis-like disorder |
X-linked hypogammaglobulinemia with hyper-IgM syndrome | Pyoderma, extensive verruca, and mucosal ulcerations |
Warts, hypogammaglobulinemia, infections and myelokathexis | Extensive verruca, skin infections, and alopecia |
Chronic granulomatous disease | Cutaneous granulomas, facial and perianal infections, aphthosis, seborrheic dermatitis, folliculitis, and neutrophilic-like disorders |
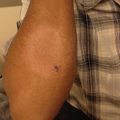
Leukocyte adhesion deficiencies | Delayed separation of umbilical stump, facial and perianal infections, poor wound healing, wounds become large and similar to pyoderma gangrenosum (Fig. 24.1), and blueberry muffin lesions in LAD Type 3 |
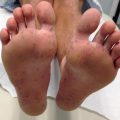
Severe combined immunodeficiency | Severe seborrheic dermatitis, erythroderma, morbilliform eruption, alopecia, candidal infections (Fig. 24.2), absent reactive lymphadenopathy, perianal rash and thrush |
Management Strategies
Patients with primary immunodeficiency disorders require a multidisciplinary approach with collaboration from immunology, infectious disease, dermatology, and others. Appropriate antimicrobial agents, and topical and systemic therapies should be administered for corresponding cutaneous manifestations, depending on the patient’s presentation (refer to the corresponding sections in this textbook for treatment of specific diseases). When indicated, surgical treatment of wounds and abscesses may be necessary. The goal of dermatologic care is to treat any active cutaneous issues, maintain the integrity of the skin barrier, and prevent trauma or other insults to the skin surface in order to avoid infection.
Investigations Recommended
For diagnosis | |
|---|---|
Test | Purpose of evaluation |
Punch biopsy of skin | Differentiate between SCID and GVHD from maternal-cell engraftment |
Culture of skin lesions or wounds | Check for bacterial, atypical mycobacterial, and fungal infections |
CBC with differential and peripheral smear, ESR | Leukocytosis in CGD and LAD; anemia in CGD and assess level of inflammation in CGD |
Flow cytometric dihydrorhodamine assay | Detects CGD (in place of the nitroblue tetrazolium test) |
Quantitative immunoglobulins | Check for immunoglobulin deficiencies; hypergammaglobulinemia in CGD |
ACE level | Rule out sarcoidosis if granuloma formation |
Referrals to: hematology/oncology and infectious disease if clinically appropriate | Evaluate for underlying disease and immunodeficiency |
Recommended Therapies
Antibody replacement for immunoglobulin deficiency | B |
Systemic steroids for development of granulomas in skin or visceral organs: | E |
Methylprednisolone 1–2 mg/kg/day with gradual taper | |
Prophylactic antimicrobial therapy: | A |
Trimethoprim and sulfamethoxazole 510 mg/kg/day divided every 12 h, three times a week, on consecutive days | |
Itraconazole therapy 5 mg/kg/day in CGD | |
Hematopoietic stem cell transplant in severe cases of CGD, LAD, SCID, and combined variable immunodeficiency | B |
In general, management of immunoglobulin deficiencies consists of replacing the deficient or absent immunoglobulin, with the exception of IgA deficiency [86]. When a deficiency is not life threatening, systemic steroids are beneficial in conditions that develop cutaneous or visceral granulomas or symptoms similar to sarcoidosis [7]. CGD requires prophylactic therapy to prevent serious bacterial or fungal infections. This is managed with daily administration of itraconazole and trimethoprim-sulfamethoxazole [43, 74, 109].
In SCID, hematopoietic stem cell transplantation is the only chance of survival, and it is most successful if performed within the first 3 months of life. It should also be considered in common variable immunodeficiency although there is a high mortality rate with the procedure [143].
Graft-Versus-Host Disease
Clinical Features
Graft-versus-host disease (GVHD) occurs when transplanted immunocompetent cells, which are introduced into an immunocompromised host, cause an immunologic insult to the host. A complex interaction occurs between the donor’s and recipient’s humoral and adaptive immunities, resulting in signs and symptoms similar to those seen in autoimmune diseases. Morbidity and mortality vary depending on the extent of involvement and the host’s response to treatment. Hematopoietic stem cell transplant or nonirradiated blood products administered to an immunocompromised patient are common associations with GVHD. Despite preventive and protective measures before, during and after the transplant, GVHD remains a significant cause of morbidity and mortality in transplant patients.
Historically, GVHD has been classified as acute or chronic, with 100 days being the cutoff time between the two. Given changes in transplant medicine and subsequent patient outcomes, the National Institutes of Health Consensus Development Project on the Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease has adapted the GVHD classification to the following: Acute GVHD includes (1) classic acute disease occurring within 100 days after transplantation and (2) persistent, recurrent, late-onset acute GVHD with an appearance of acute GVHD occurring beyond 100 days after transplantation (often seen with tapering or withdrawal of immunosuppressive therapy). Chronic GVHD includes a (1) classic chronic GVHD subtype and (2) an overlap syndrome with features of chronic and acute GVHD [42].
Early in the course of GVHD, findings may be subtle. Some patients may feel fatigue, lethargy, and mild pruritus, or develop slight erythema, all of which appear similar to a viral illness. Non-transplant physicians may help identify drug reactions, infections (especially viral exanthems in children), recurring malignancy or other skin conditions, which must be excluded prior to diagnosing GVHD.
Acute GVHD may present with pruritus, edema, erythema, and dysesthesia. As the process continues, skin involvement may range from erythematous macules to papules, vesicles, bullae, or ulceration (Figs. 24.3 and 24.4). Acute GVHD with generalized erythroderma or resembling Stevens-Johnson syndrome heralds a more severe reaction. Extracutaneous features often include cholestasis, nausea, vomiting, anorexia, diarrhea, or ileus [42]. The severity of the acute reaction is based on the skin findings, bilirubin levels, and gastrointestinal involvement.
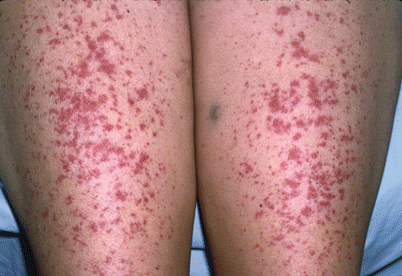
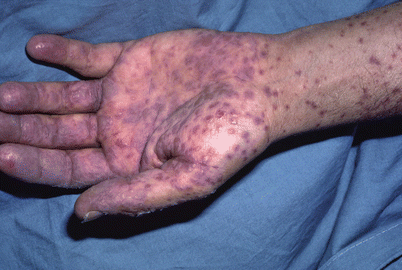
Fig. 24.3
Pupuric macules in graft-versus-host disease (Courtesy of Chauncey McHargue, MD)
Fig. 24.4
Violaceous macules and papules in graft-versus-host disease (Courtesy of Chauncey McHargue, MD)
Chronic GVHD can affect any organ system, although the skin may be the only organ involved (see Table 24.2).
Table 24.2
Cutaneous features of acute and chronic GVHD
Acute GVHD | Chronic GVHD |
|---|---|
Folliculocentric or morbilliform erythema with confluence | Diagnostic |
Edema | Poikiloderma |
Dysesthesias | Lichen planus-like |
Sclerodermatous | |
Morphea-like | |
Lichen sclerosus-like | |
Characteristic but not diagnostic | |
Keratosis pilaris-like | |
Ichthyosiform | |
Depigmentation | |
Nail dystrophy, onychorrhexis, pterygium, and nail loss | |
Alopecia, scarring or non-scarring | |
Shared features | |
Erythema | |
Morbilliform rash | |
Pruritus | |
Management Strategies
Because of the potential severity of GVHD and often sub-optimal results with treatment, the main focus is on prevention. Pretreatment of transplant tissue via elimination of immunoreactive T cells and the use of immunosuppressive medications during the first 100–180 days post-transplant attempt to prevent GVHD. Prophylactic systemic medications that are most commonly used include tacrolimus, mycophenolate mofetil, methotrexate, cyclosporine, and steroids. Additionally, bowel rest, irradiation of blood products, and prophylaxis for fungal, viral, and bacterial infections also aim to prevent GVHD.
Acute GVHD is a risk factor for developing chronic GVHD, and chronic, progressive GVHD portends a poor prognosis. Therefore, treatment of any type of GVHD is important and depends on disease severity. Topical treatments for mild cutaneous symptoms are adequate [42]. When patients have more moderate to severe disease or multi-organ involvement, systemic immunosuppression is required. Additionally, contractures seen with sclerodermoid-type GVHD may need intervention by occupational and physical therapy [29]. A multidisciplinary approach, regardless of acuity, is prudent.
While the transplant team carefully manages patients, the non-transplant physician plays an important role in disease monitoring and patient education (see Table 24.3).
Table 24.3
Recommended guidelines for non-pharmacologic evaluation and management of GVHD
Routine full-body skin evaluation performed by dermatologist every 3–6 months |
Evaluate for skin changes of acute or chronic GVHD |
Evaluate for changing skin lesions worrisome for dysplastic nevi, melanoma, and nonmelanoma skin cancers |
Evaluate for portals of entry for infection |
Evaluate/inquire about genital involvement or other mucosal surface changes |
Evaluate for contractures, other sclerodermoid changes, or changes affecting range-of-motion |
Encourage routine self skin examinations |
Parental education on early warning signs of GVHD |
Parental education on photoprotection |
Broad-spectrum sunscreen and photoprotective clothing |
Skin and nail care |
Keep nails trimmed to avoid scratching and inducing skin injury |
Daily moisturizing with emollients to maintain skin barrier |
Avoid abrasive clothing or ill-fitting shoes to prevent blistering |
Investigations Recommended
Test | Purpose of evaluation |
|---|---|
For diagnosis | |
Punch biopsy of skin | Confirm GVHD vs. other cutaneous eruption |
Culture of ulcers or non-healing wounds | Check for bacterial or fungal infection |
CBC with differential (attention to eosinophilia), CMP | Check for eosinophilia, lactate dehydrogenase levels and liver function with aminotransferases and bilirubin |
For treatment | |
Lipid panel, magnesium, and uric acid | Cyclosporine |
Blood pressure | Cyclosporine |
Referrals to: transplant team, ophthalmology, pulmonology, otolaryngology, and physical therapy/occupational therapy if clinically indicated | Evaluate for GVHD manifestations in other organs and therapy for contractures from sclerodermoid-type of GVHD |
Table 24.4
First line therapies
Topical therapies | |
Topical steroids BID prn | E |
Triamcinolone 0.1 % BID for body and hydrocortisone 1 % or 2.5 % for face, folds and groin | |
If unresponsive consider wet wraps to enhance penetration or stronger topical steroids | |
Tacrolimus BID | Ca |
Pimecrolimus daily | E |
Systemic therapies | |
Prednisone | Ba |
Prednisone + cyclosporine, alternating each medication every other day | Ba |
Mycophenolate mofetil | Ba |
Methotrexate | Ba |
Systemic tacrolimus + mycophenolate mofetil | B |
Recommended Therapies
Topical steroids are the mainstay of treatment for localized cutaneous GVHD, although there are no well-designed studies in children or adults with GVHD [32]. Tacrolimus 0.03 % or pimecrolimus 1 % may similarly be considered instead of topical steroids. When topical medicaments fail to control mild disease, patients may benefit from prednisone, although results are often suboptimal [72].
Pediatric studies show that systemic tacrolimus in combination with mycophenolate mofetil is effective in reducing the incidence of acute GVHD [87]. A Cochrane review showed that mycophenolate mofetil and methotrexate are equally efficacious, although mycophenolate mofetil had a slightly better tolerability profile [63].
Table 24.5
Second line therapies
Topical therapies | |
NB-UVB ± topical steroids | C, E |
UVA1 | Ca |
Bath PUVA (8-methoxy-psoralen in bath soak for 20 min then UVA exposure), 3 times weekly until clinical improvement then taper | E |
PUVA | Ea |
Systemic therapies | |
Etanercept + topical steroids | Ba |
One pediatric study showed NB-UVB to benefit eight out of ten patients who failed first-line therapies. Other isolated pediatric case reports show good results with NB-UVB plus topical steroids for the treatment of eczematous-type GVHD [18, 124].
For both acute and chronic forms of GVHD, UVA1 as adjunctive treatment or first-line treatment may lead to complete or partial responses without concomitant steroids [144]. Bath PUVA may also be effective while avoiding the side effects of oral photosensitizers [15, 54, 144].
Etanercept, in addition to topical steroids, in grade I (skin-only) disease have reduced the incidence of development of grades II-IV GVHD when compared to topical steroids alone [44].
Table 24.6
Third line therapies
Imatinib 100 mg/day increased to 400 mg/day | Ba |
Extracorporeal photopheresis | E |
Imatinib may serve as a treatment for steroid-refractory chronic GVHD, possibly due to its anti-inflammatory effects as seen with several fibrotic diseases. An additional benefit is that patients do not require hospitalization or long-term venous access [85]. Extracorporeal photopheresis for chronic GVHD in adults and children may improve sclerotic changes in the skin for up to 12 years after the onset of GVHD [137].
Melkersson-Rosenthal Syndrome
Clinical Features
Melkersson-Rosenthal syndrome (MRS) is a rare neuromucocutaneous disease characterized by infiltration of the skin and subcutaneous tissues with noncaseating granulomas. Patients predominately present as females in the third decade of life with an incomplete triad of fissured tongue (lingua plicata), relapsing facial paralysis, and recurrent or persistent facial edema [37] (Fig. 24.5). Children tend to present differently than adults, with unilateral facial nerve palsy as the inciting event. This can precede the edema by several months. Isolated eyelid edema as a solitary presenting sign of MRS has also been reported [100] (Fig. 24.6).
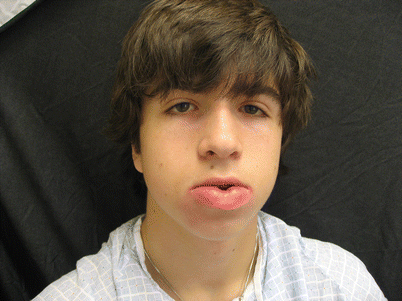

Fig. 24.5
Labial edema in Melkersson-Rosenthal syndrome (Courtesy of Tor Shwayder, MD)
Fig. 24.6
Thirteen-year-old with isolated bilateral upper eyelid edema in Melkersson-Rosenthal syndrome (Courtesy of Tor Shwayder, MD)
The etiology of MRS is unknown. It is theorized that granulomas lead to lymphatic and vascular congestion, which causes swelling of the lips, cheeks, and/or eyelids [37]. A genetic link is suspected, as up to 30 % of cases have a first- or second-degree relative with a history of “facial palsy [119].” Additionally, autoimmune diseases and infectious etiologies have been associated with MRS in some case reports.
Management Strategies
Due to the rarity of the condition and paucity of reported cases of pediatric MRS, treatment relies upon anecdotal evidence in children and studies from the adult literature. Therapeutic management is determined by the severity of disease. In solitary lip swelling, topical and intralesional therapies should be attempted first. When patients have disfiguring or debilitating symptoms, or if conservative measures are ineffective, oral agents are the next appropriate step. Finally, surgical approaches may be employed if there is permanent aesthetic and functional deformity and all other therapies have failed.
Investigations Recommended
Because of its variable presentation, MRS is a diagnosis of exclusion. It can present in a similar fashion to other disorders such as Bell’s palsy, angioedema, contact dermatitis, Crohn’s disease (CD) or infection. A skin biopsy can aid in diagnosis, but may only show edema in the early stages. Additional studies may help exclude diseases in the differential diagnosis.
Test | Purpose of evaluation | |
|---|---|---|
For diagnosis | ||
Punch biopsy of skin | Check for foreign body reaction, allergic dermatitis, lymphoma, or other granulomatous diseases | |
CBC with differential, ESR | Check for infection and assess for inflammation | |
ANA | Evaluate for underlying autoimmune disease | |
HSV PCR | HSV as etiologic agent in recurrent episodes and facial palsy | |
Complement levels (C3, C4); if abnormal, then check C1 esterase inhibitor level and function | Evaluate for angioedema | |
ACE level | Evaluate for sarcoidosis | |
Chest X-ray | Evaluate for hilar lymphadenopathy as seen in sarcoidosis | |
Patch testing | Allergic contact dermatitis to metals, foods, oral care products or other allergens | |
Referrals to: gastroenterology if clinically indicated | Evaluate for CD and possible endoscopy/colonoscopy | |
For treatment | ||
Test | Purpose | |
G6PD | Dapsone | |
CMP | Systemic medications (prednisone, methotrexate, and adalimumab) | |
PPD | Check for latent TB before using immune suppressing medications or biologics | |
Table 24.7
First line therapies
Intralesional triamcinolone 10–40 mg/ml monthly | Ea |
Topical steroid gel (fluocinonide) BID prn | Ea |
Prednisone 0.5–1 mg/kg/day | E |
Minocycline 100 mg twice daily or doxycycline 200 mg daily if permanent tooth development is complete | Ea |
Nonsteroidal anti-inflammatory drugs | Da, E |
Recommended Therapies
Few published cases of pediatric MRS are available. In the largest series to date on MRS patients with the full triad of disease, only one child was included. She responded to intralesional triamcinolone and dapsone [37]. Intralesional therapy may also help localized swelling and occasional flares [9, 117].
Systemic prednisone can be used alone or in combination with minocycline in children. Prednisone can show rapid resolution of the swelling and dramatic improvement of facial paralysis, however, rebound inflammation may occur upon its discontinuation. Minocycline has shown anti-inflammatory effects as well as inhibition of granuloma formation in other granulomatous disorders, and can be considered as an adjunctive measure with prednisone [88, 117]. After initial improvement of the symptoms, the prednisone can be tapered with gradual weaning of the minocycline, if tolerated.
Table 24.8
Second line therapies
Adalimumab | Ea |
Dapsone 125 mg once daily | E |
Methotrexate | E |
Sulfapyridine | E |
Colchicine | E |
Metronidazole 250 mg twice daily | Ea |
Tumor necrosis factor (TNF) alpha inhibitors have been used in MRS. Case reports of adults refractory to prednisone, intralesional steroids, oral antibiotics, clofazamine, methotrexate, and azathioprine had complete resolution and subsequent remission on adalimumab [104, 117]. A 10-year-old female had a complete response to dapsone plus intralesional steroids [37].
Adults with the full triad of MRS showed variable responses to dapsone, metronidazole 250 mg twice daily, and nonsteroidal anti-inflammatory drugs. Other combination therapies with reported success in the pediatric literature include: intralesional steroid injections, oral dapsone and sulfapyridine and intralesional steroid injections, colchicine, and doxycycline [71].
Table 24.9
Third line therapies
Clofazamine 100–200 mg daily; alternate dosing is 100 mg four times weekly | Da, Ea |
Plastic surgery for cheilitis granulomatosa | E |
Clofazamine is a well-known treatment of leprosy, as it has antimicrobial properties and is successful in treating granulomatous diseases. Clofazamine may decrease granulomatous infiltration of the lips in MRS, although the possible untoward side effect of skin pigmentation can occur within weeks of initiating treatment [40, 121].
Reduction cheiloplasty and facial liposuction may improve the cosmetic appearance of facial and lip swelling in treatment-resistant patients [123].
Cutaneous Crohn’s Disease
Clinical Features
Crohn’s disease (CD) is a chronic granulomatous inflammatory bowel disorder that can affect any part of the gastrointestinal (GI) system. Extraintestinal manifestations can affect the skin, joints, eyes, and liver. The etiology of CD is multifactorial and is due to an immune-mediated inflammatory process.
Pediatric patients with underlying CD most commonly present with growth failure. GI signs and symptoms may be subtle, such as mild indigestion after meals or intermittent constipation. More common manifestations of CD include diarrhea, weight loss, fever, and fatigue.
Cutaneous CD falls into one of two categories: “nonspecific” or “specific,” which are detailed in Table 24.10. About 20 % of patients with CD present with cutaneous lesions preceding the onset of GI disease by months or years. Of the children with cutaneous CD, up to two-thirds have genital involvement (Figs. 24.7 and 24.8), which may have the mistaken appearance of child abuse or sexually transmitted infections [31, 94, 98, 101]. In general, any pediatric patient with GI complaints and cutaneous lesions should be biopsied for evaluation and referred to gastroenterology. Furthermore, patients with isolated genital swelling and erythema should be evaluated for CD in the appropriate clinical setting.

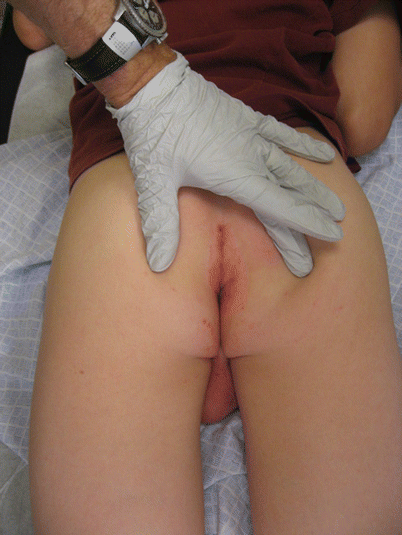
Fig. 24.7
Isolated scrotal edema in cutaneous Crohn’s disease (Courtesy of Tor Shwayder, MD)
Fig. 24.8
Perianal irritation and erythema in cutaneous Crohn’s disease (Courtesy of Tor Shwayder, MD)
Table 24.10
Cutaneous manifestation of Crohn’s disease
Specific | Nonspecific |
|---|---|
Perianal lesions Skin tags Anal fissures Fistulas Abscesses Perioral lesions Cobblestoning of mucosa Oral ulcerations Cheilitis granulomatosa Metastatic Crohn’s disease Swelling/ulceration/erythema/edema of perineum, vulva, scrotum, clitoris, labia or penis Isolated lymphedema of the genitals Sterile folliculitis Nonspecific papules, pustules, plaques, ulcers, or abscesses on any skin surface | Reactive conditions Erythema nodosum Pyoderma gangrenosum Aphthous stomatitis Erythema multiforme Epidermolysis bullosa acquisita Cutaneous vasculitis Associated conditions Vitiligo Palmoplantar pustulosis Clubbing Palmar erythema Lesions from nutritional deficiency Therapy-related Psoriasis (from TNF-α inhibitors) Lupus Drug hypersensitivities |
Management Strategies
The goal of treatment in pediatric CD is to optimize nourishment for growth and development. Additionally, management of the systemic disease by the gastroenterologist to induce remission and prevent relapse is critical. Localized lesions can be treated with topical steroids and/or intralesional steroids. If lesions are located on the face, low-potency steroids or calcineurin inhibitors should be used. Multiple lesions or complicated lesions such as fistulas or abscesses may require systemic medications [57, 96].
Finally, cutaneous or GI surgery is reserved for patients who do not respond to medications, or in patients with progressive, debilitating complications.
Investigations Recommended
Test | Purpose of evaluation |
|---|---|
For diagnosis | |
Punch biopsy of skin with special stains (acid fast and PAS) and polarized microscopy | Check for atypical mycobacteria, fungus, other granulomatous diseases or foreign body reaction |
Culture of skin lesions | Check for bacterial, atypical mycobacterial, fungal and viral causes; check for sexually transmitted infections |
CBC with differential, ESR, CRP, and CMP | Check for underlying disease and assess level of inflammation |
Chest x-ray | Evaluate for hilar lymphadenopathy as seen in sarcoidosis; evaluate for tuberculosis |
PPD | Check for TB as a cause of granulomatous disease |
Referral to: gastroenterology if CD is clinically suspected | Evaluate for CD and possible endoscopy/colonoscopy |
For treatment | |
TPMT | Azathioprine or mercaptopurine |
PPD | Check for latent TB before using immune suppressing medications or biologics |
Table 24.11
First line therapies
Systemic induction therapy for systemic CD and severe cutaneous disease | |
For mild CD | |
Aminosalicylates: | B |
Mesalamine 50–80 mg/kg/day | |
Sulfasalazine | |
Thiopurines | B |
Azathioprine 2–3 mg/kg/day | |
Mercaptopurine 1–1.5 mg/kg/day | |
For moderate to severe CD | |
Systemic steroids | B |
Prednisolone 1–2 mg/kg/day | B |
Prednisone 20 mg daily | B |
Oral budesonide 3 mg three times a day | |
For severe CD | |
Infliximab 5 mg/kg at weeks 0, 2, 6, and every 8 weeks | B |
Adalimumab | B |
Induction dosing at week 0 and 4: | |
<40 kg: 160 mg, 80 mg | |
≥40 kg: 80 mg, 40 mg | |
Systemic maintenance therapy | |
For mild CD | |
Aminosalicylates: | B |
Mesalamine 50–80 mg/kg/day | |
Sulfasalazine, max dose 4–6 g/day + folate 1 mg daily | |
For moderate to severe CD | |
Thiopurines | B |
Azathioprine 2–3 mg/kg/day | |
Mercaptopurine 1–1.5 mg/kg/day | |
Infliximab 5 mg/kg every 8 weeks | B |
Methotrexate 15 mg/m2 weekly | B |
Adalimumab | B |
Maintenance dosing every other week: | |
<40 kg: 10–20 mg | |
≥40 kg: 20–40 mg | |
Topical and additional systemic therapy for cutaneous CD | |
Tacrolimus 0.03 % ointment twice daily | D, Ea |
Intralesional corticosteroid | D |
1 ml of 10 mg/ml triamcinolone + 1 ml lidocaine 1 % for pain relief | |
Topical steroids | Ea |
Mid-high potency twice daily | |
Recommended Therapies
For induction therapy, aminosalicylates, glucocorticoids, biologics, and thiopurines are chosen, depending on the severity of the disease. For moderate to severe disease, glucocorticoids remain the first-line treatment for acute therapy. Corticosteroids are then bridged with an immunomodulator or biologic therapy to avoid side effects in the growing child [69]. Induction therapy with infliximab for moderate to severe CD had lower recurrence rates at 3 years when compared to induction with systemic steroids [68]. Adalimumab is approved for children older than 6 years of age with moderate to severe CD that is unresponsive to conventional therapies.
For the maintenance phase of treatment, many of the aforementioned therapies can be continued for control of the disease, with the exception of corticosteroids. Steroids should be reserved for acute exacerbations and tapered if there is adequate control with steroid-sparing agents. Methotrexate can also be considered a maintenance therapy alone or in combination with other medications. Furthermore, methotrexate should be considered in patients who do not respond to, or cannot tolerate, aminosalicylates and thiopurines [120, 136].
For topical treatment of cutaneous CD, tacrolimus ointment showed promising results in small series of pediatric patients and in adult case reports [28, 110]. Similarly, topical steroids and intralesional triamcinolone can be beneficial [31, 134].
Table 24.12
Second line therapies
Systemic therapy | |
Mycophenolate mofetil 15 mg/kg/day + prednisone taper | Ba |
Topical and additional systemic therapy for cutaneous Crohn’s disease | |
Metronidazole 250 mg three times a day | E |
Mycophenolate mofetil is becoming more widely utilized in CD. In cases where azathioprine is contraindicated, mycophenolate mofetil in addition to prednisone showed efficacy in inducing remission and sustaining control of the disease [82].
For cutaneous lesions unaccompanied by active gastrointestinal CD, metronidazole can also be used alone or in conjunction with methotrexate, prednisone, and topical therapies [94].
Table 24.13
Third line therapies
Systemic therapy | |
|---|---|
Thalidomide 50 mg nightly increased stepwise to 150 mg if needed | D |
Surgery | E |
Hyperbaric oxygen | Ea |
Sarcoidosis
Clinical Features
Sarcoidosis is a multisystem disorder characterized by noncaseating granuloma formation due to an unknown etiology. Although it most commonly affects adults, there are two observed pediatric forms, early-onset sarcoidosis (EOS) and a later-onset disease. The two groups vary in presentation and systemic involvement (Table 24.14).
Table 24.14
Characteristics of pediatric sarcoidosis
Characteristics | Early-onset | Later-onset |
|---|---|---|
Ages | 0–5 years | >5 years |
Etiology | NOD2/CARD15 | Unknown |
Classic presentation | Triad of arthritis, uveitis and dermatitis | Constitutional symptoms and lung involvement |
Organ involvement | Polyarticular arthritis | Fever, lethargy, malaise, cough, and dyspnea |
Uveitis (77 %) with anterior uveitis being the most common and most serious | Lacrimal glands infiltration | |
Liver, kidney, gastrointestinal tract, brain and bone | Anterior uveitis | |
Lymphadenopathy | ||
Arthritis | ||
Hematologic abnormalities | ||
Pulmonary findings | Pneumonitis | Advanced parenchymal disease |
Bronchial granulomas | Hilar lymphadenopathy | |
Reported cutaneous findings | Tan or erythematous scaly papules, plaques and/or nodules | Papules, patches, plaques, nodules, ulcerations and necrotic lesions |
Solitary growths mimicking cutaneous histiocytosis | Associated exanthem | |
Erythema nodosum | Vasculitides | |
Lesions that resolve with pitted scarring | Erythema nodosum | |
Keloidal changes to preexisting scars |
EOS is caused by a sporadic mutation in the NOD2 gene, also known as CARD15. Blau syndrome, clinically identical to EOS, is also caused by a NOD2 mutation, but is due to an autosomal dominant inheritance [27]. The course of EOS is unpredictable, as some lesions can persist for many years while others can occur intermittently with sporadic resolution. Pediatric patients in the older population (ages >5 years) tend to have advanced disease demonstrating constitutional symptoms and pulmonary findings similar to adults [147]. The cutaneous findings of the two variants of pediatric sarcoidosis have overlapping features and a wide range of distinct primary lesions (Fig. 24.9 and Table 24.14).
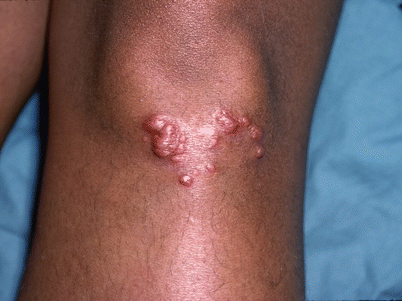
Fig. 24.9
Firm papules on the knee of cutaneous sarcoidosis (Courtesy of Chauncey McHargue, MD)
The prognosis of pediatric sarcoidosis relies upon the degree of organ involvement, as neurosarcoidosis and extensive hilar involvement have shown increased morbidity and mortality [112]. Erythema nodosum portends a favorable prognosis, similar to that seen in adults [114]. Cutaneous sarcoidosis may herald future systemic involvement, which can lag behind the cutaneous presentation by several years. Thus, even with resolution of cutaneous sarcoidosis, patients should be monitored annually for development of extracutaneous involvement [83]. While the majority of pediatric cases resolve spontaneously within 6 years, some have persistent organ damage [79].
Management Strategies
In general, treatment should reflect the activity and symptomology of disease and the type of organ involvement. If the disease is symptomatic, rapidly progressive, scarring, or otherwise affecting quality of life, the provider must address the risks and benefits of choosing a particular therapy. Limited cutaneous disease may only require topical therapy, whereas more aggressive disease often necessitates systemic therapy. Also, combinations of therapies may help control the inflammatory process.
Investigations Recommended
Sarcoidosis is a diagnosis of exclusion. It is important to check inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein. Angiotensin converting enzyme levels are helpful in the older pediatric population due to the involvement of the lung as well as its utility in following disease activity and response to treatment. However, it must be noted that unaffected children can have angiotensin converting enzyme levels greater than two standard deviations of the adult range [103].
Test | Purpose of evaluation |
|---|---|
For diagnosis | |
Skin punch biopsy with PAS or biopsy of lymph node with PAS if no skin lesions | Check for other granulomatous diseases |
Culture of blood, stool, urine, and cerebrospinal fluid | Check for fungal, bacterial or viral causes of granulomatous disease |
CBC with differential, CMP, CRP, and ESR | Check for hypercalcemia, organ involvement, and assess level of inflammation |
ANA | Evaluate for underlying autoimmune disease |
Rheumatoid factor | Rule out rheumatoid arthritis |
Urinalysis | Monitor kidney function and check for proteinuria |
1,25-dihydroxy vitamin D | Increased levels can be seen in sarcoidosis causing hypercalcemia |
ACE level | Level of disease activity in sarcoidosis |
Chest X-ray and joint X-rays | Evaluate for joint deformities/swelling |
Referrals to: rheumatology, ophthalmology, pulmonology, nephrology, and cardiology if clinically indicated | Evaluate for multiorgan disease; ophthalmology to evaluate for use of antimalarials; cardiology to evaluate for electrocardiogram for conduction defects due to granulomas |
For treatment | |
TPMT | Azathioprine |
ACE level | Therapeutic monitoring |
PPD | Check for latent TB before using immune suppressing medications or biologics |
Table 24.15




First line therapies
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
