2 Craniofacial Syndromes
Summary
The diagnosis and management of syndromic craniofacial anomalies, namely Apert, Crouzon, Muenke, Pfeiffer, Saethre-Chotzen, and Treacher Collins syndromes, are complex due to the wide phenotypic variability of the clinical presentations. Craniofacial syndromes can be divided into two major categories: craniosynostosis-associated syndromes and cleft-associated syndromes. Common syndromic craniosynostosis include Apert, Crouzon, Muenke, Pfeiffer, and Saethre-Chotzen, while Treacher Collins is the most common cleft-associated mandibulofacial dysostosis condition. Management of patients with craniofacial syndromes includes correctly timed surgical operations in a staged fashion, close involvement of the patients’ families, as well as psychosocial and aesthetic considerations for the pediatric patients. A multidisciplinary team approach is vital to a successful outcome, and the involvement of a pediatric plastic surgeon in the surgical management of these complex craniofacial syndromes is central.
2.1 Introduction
Craniofacial syndromes can be divided into two broad categories: craniosynostosis-associated syndromes and cleft-associated syndromes. The clinical diagnosis and surgical management of craniofacial syndromes, such as Apert’s, Crouzon’s, Muenke’s, Pfeiffer’s, Saethre–Chotzen, and Treacher Collins syndromes, are complex due to the overlapping and varied phenotypic presentations and genetic mutations of the different syndromes. For example, all craniosynostosis syndromes have abnormal, premature fusion of one or more cranial sutures. However, some syndromes, such as Apert’s and Pfeiffer’s syndromes, have prominent midface extrusion, whereas the midface position is less severely affected in other conditions, such as Muenke’s and Saethre–Chotzen syndromes. Thus, correct diagnosis and treatment for these conditions require a multidisciplinary team approach. Surgical and nonsurgical management needs to be tailored based not only on the genetic and clinical diagnosis but also on the severity of symptoms and functional compromise experienced by the patient. Optimal treatment quite often necessitates multiple surgical disciplines, with the involvement of a pediatric plastic surgeon being essential for a successful outcome.
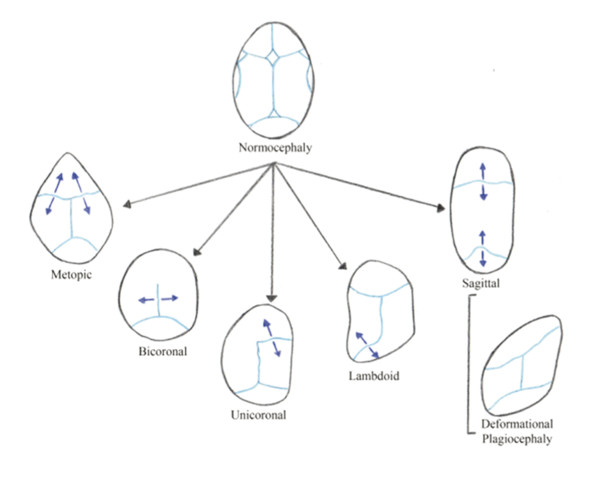
Craniosynostosis results from the premature fusion of one or more cranial sutures and is a relatively common congenital defect, affecting 1 in every 2,000 births globally. Premature fusion restricts the growth of the skull perpendicular to the suture involved; consequently, compensatory skull growth occurs parallel to the affected suture, in order to accommodate the growing brain (Fig. 2‑1). Craniosynostosis usually occurs as an isolated condition but can also result from a detectable genetic mutation. Apert’s, Crouzon’s, Muenke’s, Pfeiffer’s, and Saethre–Chotzen syndromes are unique for their designation as syndromic craniosynostoses and are collectively the most common syndromic craniosynostoses. More than 100 syndromes have been described and attributed to specific genetic mutations, notably fibroblast growth factor receptor-1 (FGFR-1), FGFR-2, and FGFR-3, and the transcription factors TWIST (upstream regulator of FGFRs) and MSX-2. Specifically, gain-of-function mutations are associated with the FGFR genes and human MSX-2, whereas loss-of-function mutations are associated with the TWIST gene. Mutations in FGFR-2 gene are the most prevalent cause for three notable, overlapping syndromes: Apert’s, Crouzon’s, and Pfeiffer’s syndromes (Table 2‑1).
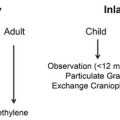
Surgical intervention remains the only treatment to correct the underlying pathology. The main goals of surgery are expansion and normalization of the calvarial shape to increase the intracranial volume and normalization of craniofacial proportions and occlusal relationships. Early (<1 year of age) procedures aim to allow adequate intracranial volume for brain growth and avoid potential consequences of increased intracranial pressure. Surgical procedures during childhood and teenage years aim to correct midface retrusion and to improve the airway, bony orbital volume, and occlusion. The two main surgical procedures for craniosynostosis repair are fronto-orbital advancement (FOA) with open cranial vault remodeling and strip craniectomy, which can be performed in a minimally invasive endoscopic-assisted fashion. Each approach has its own advantages and disadvantages and may or may not be indicated based on the type of craniosynostosis and the age of presentation. Complications associated with surgical correction include bleeding, infection, optic nerve ischemia, seizures, delayed healing and other wound problems, incomplete bone healing resulting in chronic bone defects, contour irregularities, and need for additional surgical procedures.
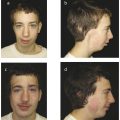
Treacher Collins syndrome differs from the aforementioned syndromes in that it is not a syndromic craniosynostosis. By contrast, Treacher Collins syndrome is the most common cleft-associated syndrome and is also known as mandibulofacial dysostosis. Occurring in 1 in 50,000 live births, Treacher Collins syndrome is an autosomal dominant disorder of craniofacial development, and its major clinical features include bilateral absence or hypoplasia of the zygomas, cleft palate, midface hypoplasia, micrognathia, microtia, and conductive hearing loss. Recently, mutations in TCOF1 (Treacher Collins–Franceschetti syndrome 1) and POLR1D, in chromosome 5, have been identified as the cause of Treacher Collins syndrome. The goals of surgical treatment and overall management of patients with Treacher Collins syndrome include palate repair, speech therapy, construction of ears, reconstruction of the zygomatic prominence, and correction of midface retrusion to improve the airway and respiratory status as well as to improve occlusion. Treacher Collins syndrome highlights and provides an excellent example of the complex, surgically staged, multidisciplinary team approach requisite for the care of patients with congenital disorders, particularly those with cleft palate.
Herein, we describe the clinical diagnosis and management of six syndromes, Apert’s, Crouzon’s, Muenke’s, Pfeiffer’s, Saethre–Chotzen, and Treacher Collins syndromes, which together are reflective of the wide spectrum of craniofacial syndromes confronting pediatric plastic and reconstructive surgeons today.
2.2 Diagnosis
2.2.1 Craniosynostosis-Associated Syndromes
Apert’s syndrome, or acrocephalosyndactyly type I, is seen in 1 in 65,000 live births and accounts for 4.5% of all craniosynostosis cases. Characteristic clinical features of Apert’s syndrome are craniosynostosis (usually bicoronal), turribrachycephaly, down-slanting palpebral fissures, hypertelorism, exorbitism, cleft palate, midface hypoplasia or retrusion, and complex syndactyly of the hands and feet. The syndrome is caused by one of the two FGFR-2 mutations on chromosome 10 (10q26.13), Ser-252 and Pro-253. It either is inherited in an autosomal dominant fashion or can arise de novo, with older paternal age being a recognized risk factor for the de novo mutation. A specific missense substitution involving adjacent amino acids, Ser-252-Trp and Pro-253-Arg, in the linker between the second and third extracellular immunoglobulin domains of FGFR-2 has been identified as the causal mutation resulting in Apert’s syndrome. Interestingly, there is a varying genotype–phenotype correlation among the two noted FGFR-2 mutations, Ser-252 and Pro-253. Cleft palate, nasolacrimal stenosis, and severe ocular findings, such as ptosis, strabismus, and amblyopia, are more common in patients with the Ser-252 mutation in FGFR-2. However, the degree of syndactyly and intellectual disability is more significant in patients with the Pro-253 mutation in FGFR-2. Moreover, males and females are affected equally, and 50% of children with a parent diagnosed with Apert’s syndrome will inherit the condition. To confirm the diagnosis in a proband, targeted mutation analysis of FGFR-2 for the mutations Ser-252-Trp and Pro-253-Arg should be conducted, and if normal, sequence analysis for rare FGFR-2 mutations or partial gene insertions or deletions should be considered. In terms of embryological etiology, Apert’s syndrome is classified as a brachial arch syndrome affecting the first brachial arch, which is the precursor to the maxilla and mandible.
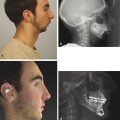
The two most notable clinical features of Apert’s syndrome are craniosynostosis and severe syndactyly of the hands and feet (Fig. 2‑2 and Fig. 2‑3). The Apert’s hand is a distinctive and pathognomonic feature of the syndrome and is well described by the Upton’s classification (Table 2‑2). As Upton specified, the four most distinguishing features of the Apert’s hand are as follows: (1) a short thumb with radial deviation, (2) complex syndactyly of the index, long, and ring finger, (3) symbrachyphalangism, and (4) simple syndactyly of the fourth web space. In fact, Apert’s syndrome is most easily clinically differentiated from Crouzon’s syndrome by the soft tissue and bony syndactyly in the hands and feet, which are not seen in Crouzon’s syndrome. In addition, the facial and cranial features of Apert’s syndrome are more prominent at birth than with Crouzon’s syndrome, in which features develop progressively throughout infancy.
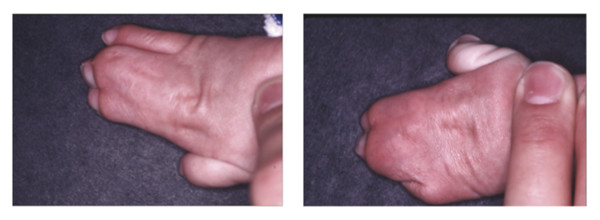
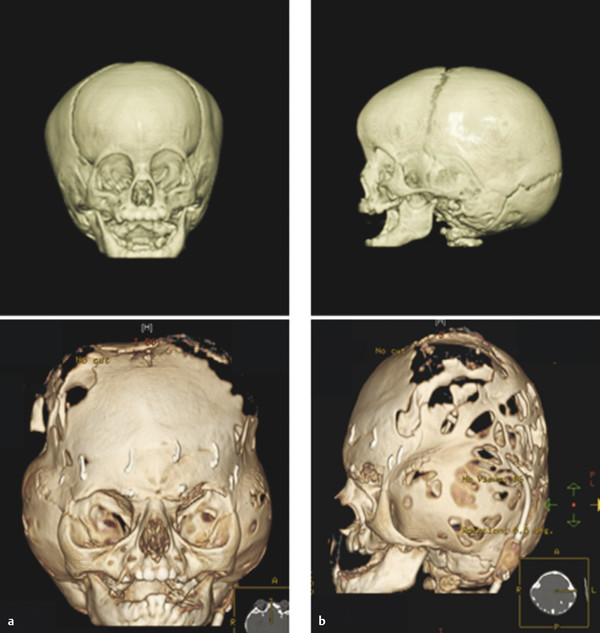
Other cephalometric diagnostic features of Apert’s syndrome include a shortened and widened anterior cranial fossa and reduced bony orbital volume, leading to exorbitism. In the midsagittal plane, there is a wide-open calvarial defect from the root of the nose to the posterior fontanelle. Cloverleaf skull, or kleeblattschadel, has been rarely reported in association with Apert’s syndrome. There is a high incidence of cleft palate at 50%, divided equally with 25% true clefts and 2% pseudo-clefts. The majority of patients with Apert’s syndrome have a high-arched palate, which is seen in conjunction with an anterior open bite and dental overcrowding. Notably, there is no association with cleft lip. The lips are crossbow-shaped or trapezoidal, and the lower lip usually protrudes. Ocular findings include possible globe subluxation, downward slant of the lateral canthi, and a well-recognized S-shaped upper-eyelid ptosis. Finally, there is often significant cutaneous involvement, with abnormally thick skin encapsulating skeletal abnormalities, seborrhea, and eventually facial acne as the patient ages.
Sequelae of Apert’s syndrome include, but are not limited to, poor intellectual development, obstructive sleep apnea (OSA), repeated ear or sinus infections, hearing loss, and ophthalmic morbidity, namely strabismus with more esotropia than exotropia.
Crouzon’s syndrome, or craniofacial dysostosis type I, has an incidence of 1 in 60,000 live births in the United States (1 in 25,000 globally) and accounts for 4.8% of all craniosynostosis cases. The syndrome is caused by a mutation in FGFR-2, and the specific gene is on chromosome 10 (10q26.13). A specific mutation within the FGFR-3 gene accounts for Crouzon’s syndrome and acanthosis nigricans. Similar to Apert’s syndrome, Crouzon’s syndrome is an autosomal dominant genetic disorder as well as a syndrome affecting the first brachial arch, which is the precursor for the maxilla and mandible. A tall, flattened forehead secondary to usually bicoronal craniosynostosis, a beaked nose, exorbitism, and mid-face hypoplasia characterize the syndrome. Usually, Crouzon’s syndrome has a milder degree of craniofacial deformity clinically when compared with Apert’s syndrome. In contrast to Apert’s syndrome, patients with Crouzon’s syndrome rarely have cleft palate and usually have structurally normal hands and feet as well as relatively normal intelligence.
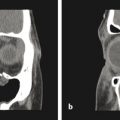
Diagnosis of Crouzon’s syndrome is most often made by clinical assessment in infancy. Clinical suspicion is heightened when the common triad of craniosynostosis, midfacial hypoplasia, and exorbitism is recognized. Further confirmatory studies include genetic testing, skull radiographs, magnetic resonance imaging scans, and computed tomographic scans. Craniosynostosis is usually bicoronal, and in many cases, the synostosis can be progressive in patients with Crouzon’s syndrome. The craniosynostosis of Crouzon’s syndrome is typically associated with a distinctive shortening of the anterior cranial fossa depth, with orbital roof hypoplasia and advancement of the posterior wall of the orbits, thus resulting in a severe ocular proptosis and shallow orbits. Cervical abnormalities may be part of the syndrome and can include butterfly vertebrae and fusions of posterior elements. In addition, features of Crouzon’s syndrome include conductive hearing loss, strabismus, anterior overbite, short upper lip, mandibular prognathism, hypoplastic maxilla, and small nasopharynx, necessitating mouth breathing.
Muenke’s syndrome has an incidence of 1 in 30,000 live births and is the most common form of syndromic craniosynostosis, accounting for up to 8% of all cases of craniosynostosis. The syndrome is autosomal dominant and is due to a mutation on the FGFR-3 gene, specifically a Pro-250-Arg base substitution mutation that is found in 100% of the patients with the syndrome. It was the first clinical syndrome that was defined first on a molecular basis by genetic mutation.
Clinically, Muenke’s syndrome presents with craniosynostosis, usually bicoronal (71% of patients) or unicoronal (29% of patients), and can occasionally present with megalocephaly, without craniosynostosis. Patients have varying degrees of midface hypoplasia, with ocular hypertelorism and strabismus. In contrast to Apert’s syndrome, patients with Muenke’s syndrome usually have normal hand and foot phenotypes but can reportedly be found to have carpal or tarsal fusions. Unlike the hearing loss characteristic of other syndromic craniosynostosis, patients with Muenke’s syndrome suffer from sensorineural hearing loss. Finally, developmental delay and intellectual disabilities have been reported in 33% of patients with Muenke’s syndrome.
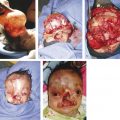
Pfeiffer’s syndrome, or acrocephalosyndactyly type V, has an incidence of 1 in 100,000 live births and is notable among the group of syndromic craniosynostosis for its high mortality and need for multiple surgical interventions (Fig. 2‑4). Genetically, the syndrome is autosomal dominant, with incomplete penetrance and variable expressivity, and is linked to two genes FGFR-1 (8p11.23–p11.22) and FGFR-2 (10q26.13). However, unlike Muenke’s syndrome, where there is a one-to-one genotype–phenotype correlation for the syndrome, Pfeiffer’s syndrome boasts a wide spectrum of causal mutations and genotypic variability; thus, the genetic diagnosis should be approached systematically. To confirm the diagnosis of Pfeiffer’s syndrome, sequence analysis of exons 8 and 10 should be done, since these are the locations for approximately 80% of FGFR-2 mutations. If normal, then sequence analysis of exons 3, 5, 11, 14, 16, and 17 should be pursued, since these are the sites of 10% of FGFR-2 mutations. Testing of FGFR-1 mutation should be considered if an FGFR-2 mutation is not found or if the patient has a milder clinical phenotype.
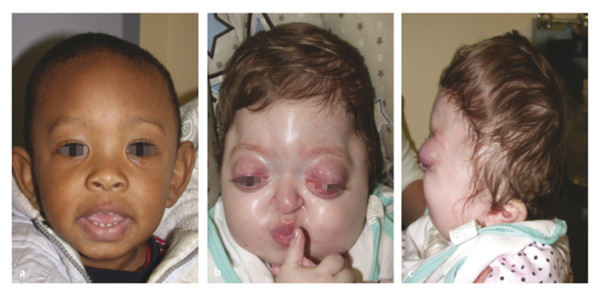
Clinically, broad thumbs and big toes, valgus deformities, and, occasionally, cardiac abnormalities distinguish patients with Pfeiffer’s syndrome from those with other syndromic craniosynostosis. In fact, measuring the angles between extended digits of the hands can facilitate a clinical diagnosis of Pfeiffer’s syndrome. In addition, synostoses of the elbow and knee, hydrocephalus, and imperforate anus are reported frequently as associated with Pfeiffer’s syndrome.
Owing to the wide phenotypic and genotypic variability of the syndrome, Pfeiffer’s syndrome has been further classified into three types according to a classification system described by Cohen et al (Table 2‑3). Pfeiffer’s syndrome type I involves the classic, common phenotype of symmetric bicoronal craniosynostosis, variable syndactyly, broad thumbs, widened great toes, normal intelligence, and relatively normal life span (or rather survival to adulthood). Pfeiffer’s syndrome type II has characteristic findings of severe ocular proptosis, ankylosis of the elbows, broad thumbs, widened great toes, severe central nervous system involvement with hydrocephalus, and visceral anomalies and carries a poor prognosis. Pfeiffer’s syndrome types II and III have significantly more severe phenotypes, with bicoronal craniosynostosis in addition to multiple other sutures, as well as severe developmental delay and shortened life spans. Of note, the cloverleaf skull anomaly (kleeblattschadel) is unique to Pfeiffer’s syndrome type II.
Saethre–Chotzen syndrome, or acrocephalosyndactyly type III, has an incidence in the range of 1 in 25,000 to 1 in 50,000 live births, and it is one of the most common types of syndromic craniosynostosis. The syndrome is autosomal dominant, with high penetrance and variable expressivity, involving mutations of the TWIST1 gene on chromosome 7 (7p21.1).
Clinically, classic diagnostic findings for Saethre–Chotzen syndrome include unilateral or bilateral coronal craniosynostosis, strabismus, ptosis, low frontal hairline, and malformed ears, each with a distinctly small pinna and prominent superior crus. In contrast to Apert’s syndrome, patients with Saethre–Chotzen syndrome neither have syndactyly features of the hands and feet nor have the proptosis feature characteristic of Crouzon’s syndrome. Patients with Saethre–Chotzen syndrome are clinically differentiated by their low-set frontal hairline, parrot-beaked nose, brachydactyly, and milder bone deformities. In addition, patients may have partial syndactyly of the fingers, usually between the web space of the index finger and long finger.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


