6 Rare Craniofacial Clefts
Summary
The treatment of rare craniofacial clefts starts with making an accurate diagnosis, including an identification of all altered or missing facial soft tissue and skeletal structures.
The ordered Tessier classification system facilitates in this diagnostic process and in surgical planning.
Adherence to basic, established surgical principles of soft tissue and skeletal reconstruction facilitate treatment of even the most complex composite defects.
Surgical intervention is often staged and carried out over many years to achieve a desirable end result.
The best approach to the longitudinal care of patients with rare clefts is through close collaborative multidisciplinary input.
6.1 Introduction
“In the midst of chaos there is also opportunity” — Sun Tzu. As surgeons, if we look for “common presentations of common diseases” to facilitate our attempts to render accurate diagnoses, develop appropriate treatment plans, and achieve predictable surgical outcomes, then the spectrum of disorders collectively referred to as the rare craniofacial clefts can frustrate even the most skilled and resolute among us. The rare craniofacial clefts represent aberrations of development that challenge our diagnostic skills and our technical abilities. However, like any other complex surgical disease, the key to successful management is making an accurate assessment of the problem, developing a specific set of longitudinal surgical goals, and having an appreciation for how sequential surgical results will evolve over time.
6.2 Classification and Diagnosis
6.2.1 Embryology
Although a comprehensive discussion of human craniofacial embryology is well beyond the scope of this chapter, the complexity of the rare facial clefts and their derivation from failures of embryogenesis merit a brief review of the relevant aspects of in utero facial development.
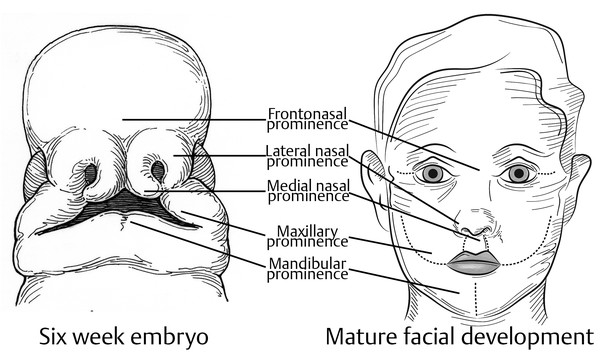
Much of the critical development of the craniofacial skeleton occurs between the fourth and eighth weeks of gestation, when the endoderm, mesoderm, and ectoderm layers of the face migrate toward the midline. The frontonasal, maxillary, and mandibular processes follow programmed patterns of migration and fusion to create balanced facial features, including the cranial base, forehead, orbits, zygomas, maxilla, nose, stoma, and mandible (Fig. 6‑1). Whether due to a failure of fusion of these migrating processes or due to insufficient migration of mesenchymal cells into the planes of tissue fusion at the leading margins of these processes—or a combination of both—errors in embryogenesis involving these structures can manifest as vertical, oblique, or transverse clefts.
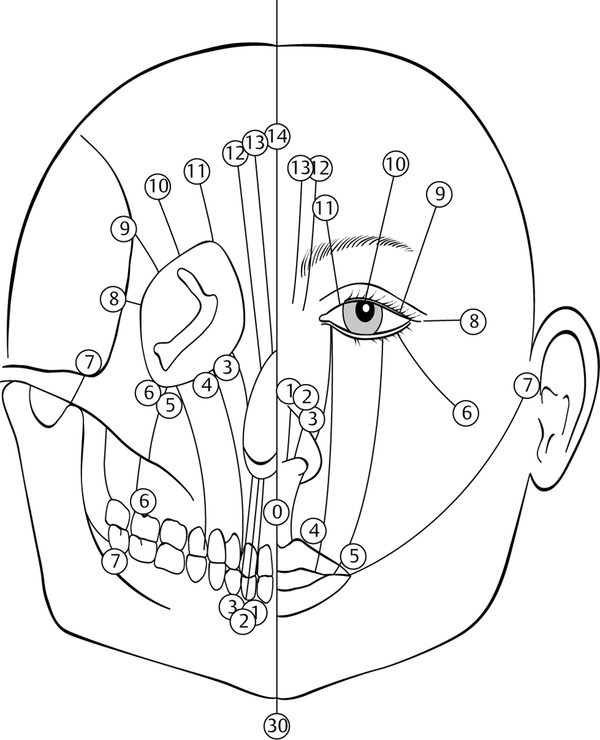
When Paul Tessier presented his approach to classifying rare craniofacial clefts to the second International Congress on Cleft Palate in Copenhagen in 1973, he offered a comprehensive and concise way to categorize not only the obvious soft tissue deformities but also the underlying aberrant bony anatomy. His logical approach to a set of deformities that seemingly defy logic has since helped us accurately diagnose and appropriately manage these patients.
Dr. Tessier divided the face into various “zones” that are centered on imaginary axes running horizontally through the orbits and vertically through the facial midline. Soft tissue and bony clefts occurring below the orbits are numbered 0 through 8 and those above the orbits are numbered 9 through 14. This numbering system encourages us to evaluate the entire height of the face and thereby more readily identify coincident facial and cranial clefts. When this occurs, the numbers of the individual clefts add up to 14 (e.g., 0–14, 1–13, and 2–12 clefts). Interestingly, Tessier defined clefts of the lower lip, mandible, and associated midline soft tissues of the lower face and neck as number 30 clefts (Fig. 6‑2).
These clefts may be characterized by tissue excess or deficiency; may involve soft tissue, bone, or both; may be unilateral or bilateral; and may be associated with numerous other clefts of diverse orientation. Deconstructing complicated and overlapping cleft patterns according to the diagnostic hallmarks of the individual component clefts, as described later, helps establish accurate diagnoses and develop appropriate treatment plans. In general, physical examination should be complemented by computed tomography (CT) to identify underlying bony abnormalities and by magnetic resonance imaging (MRI) to define the extent of intracranial disease, if appropriate.
Number 0 Cleft
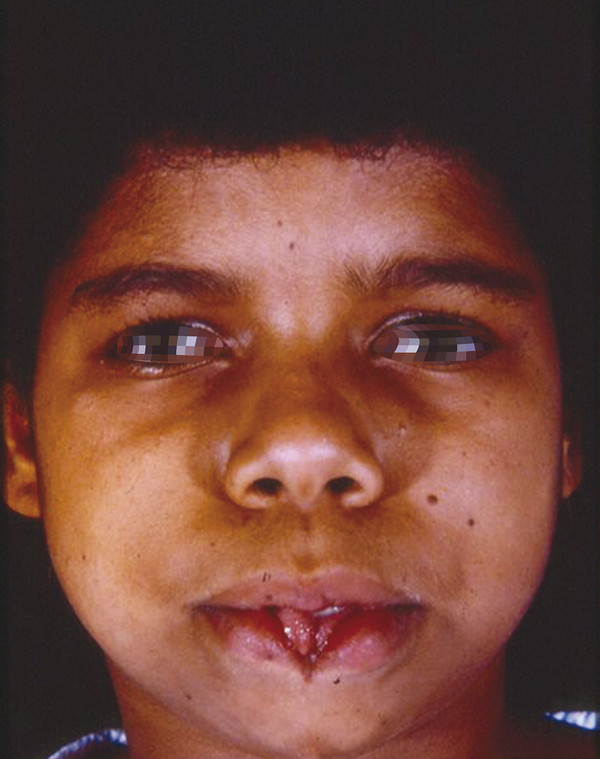
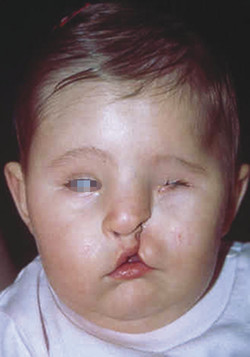
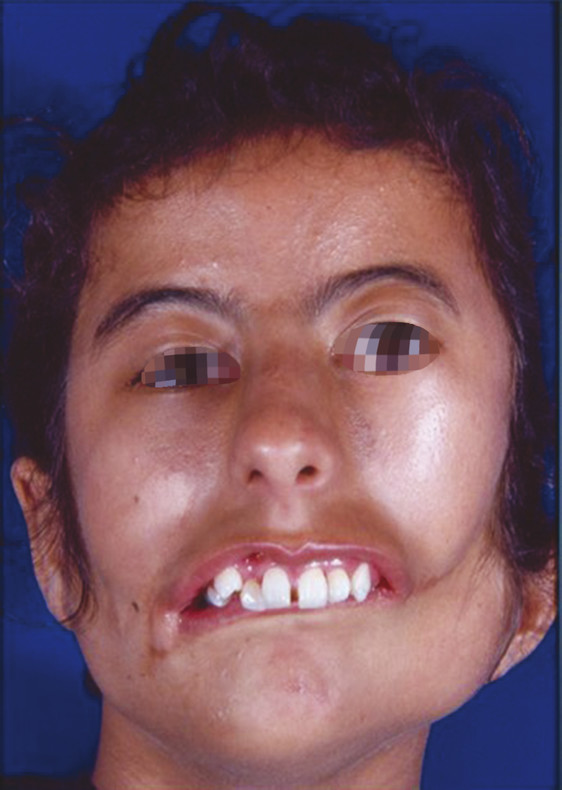
The zero cleft is a midline defect that may involve soft tissue and bone from the central incisors up through the nasal cavity and the perpendicular plate of the ethmoid bone. Mild presentations may be limited to an isolated midline cleft of the lip. With progressive severity, one may note a high and arched or complete cleft palate with absent premaxilla and philtrum, central cleft of the nose with absent columella and nasal septum, hypoplasia of the nasoethmoid complex, arrhinia, and hypotelorbitism. Alternatively, midline tissue excess may result in enlarged and broadened nasal bones, widened nasal septum, and enlarged and laterally or superiorly displaced nasal cartilages. The isolated zero cleft is very rare; it is usually associated with the number 14 Tessier cleft (Fig. 6‑3).
Number 1 Cleft
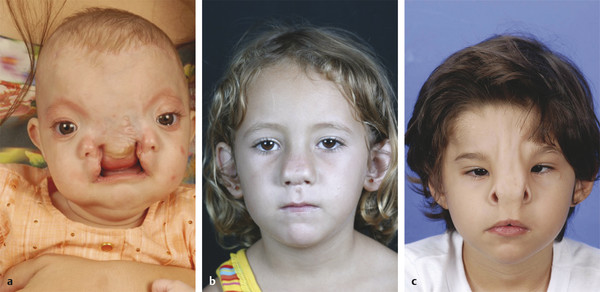
The number 1 cleft is a vertical paramedian cleft characterized by a cleft lip in the region of the Cupid’s bow and soft tissue notching through the dome of the nose that extends toward the medial canthus and medial brow. The nasal defect may include soft tissue fissures over the nasal dorsum or completely missing upper and lower lateral cartilages. Heminasal atrophy or a proboscis may be seen in severe forms. A bony cleft between the central and lateral incisors may extend into the pyriform aperture and cephalad to involve the ethmoid sinuses, the nasal bone, and the frontal process of the maxilla, resulting in hypertelorbitism (Fig. 6‑4).
Number 2 Cleft
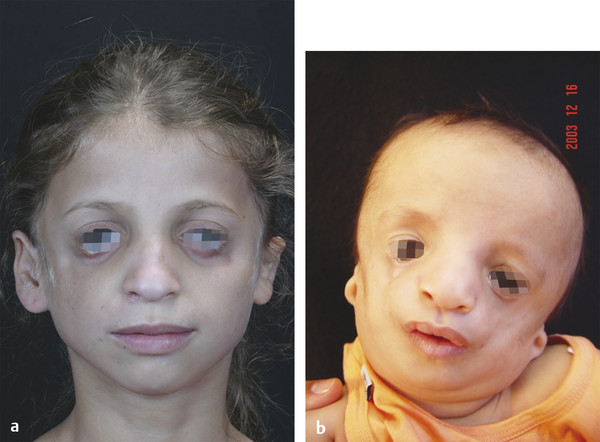
In its isolated form, the number 2 cleft is very rare. The cleft lip, such as the number 1 cleft, occurs in the area of the Cupid’s bow. The nose is characterized by flattening of the medial third of the nostril, without true notching. The nasal dorsum may be widened, and the septum may be deviated. The palpebral fissure and medial brow are intact but may exhibit inferior displacement or epicanthal folding. The bony cleft begins in the region of the lateral incisor, and the skeletal fissure extends cephalad into pyriform aperture. The frontal process of maxilla is broad and flat and can be notched. Ethmoid enlargement contributes to hypertelorbitism.
Number 3 Cleft
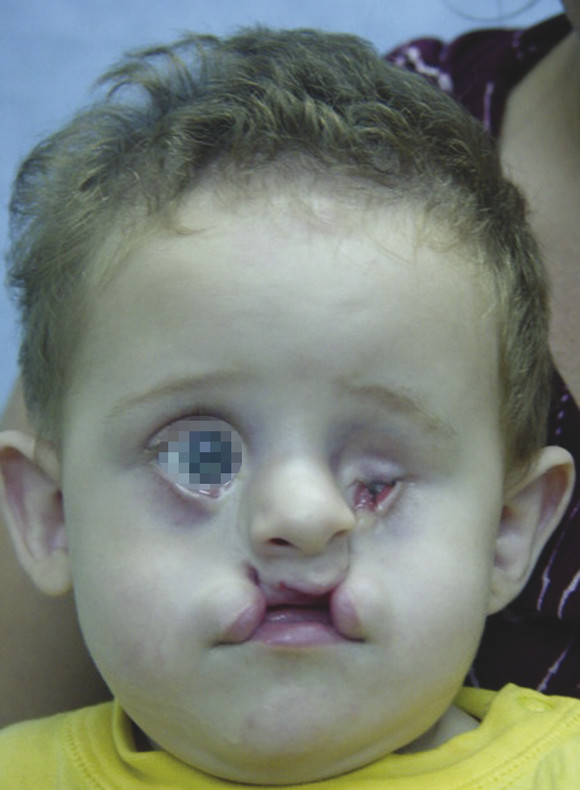
The number 3 cleft has been described as an oblique cleft, due to the vector of tissue involvement, and as an oro-naso-ocular cleft, due to the facial cavities that it might involve. The soft tissue cleft again begins in the area of the common cleft lip. It continues along the alar base and then obliquely cephalad toward the medial canthus. The cheek tissue deficiency foreshortens the distance from the nasal ala to the medial canthus, causing inferior displacement of this structure. A lower-eyelid coloboma medial to lacrimal punctum may be present, along with agenesis of part or all of nasolacrimal system. Microphthalmia, anophthalmia, and epibulbar dermoids may be seen in this cleft. The cleft in the alveolus occurs between the lateral incisor and the cuspid and extends through the lateral border of the pyriform into the nasal cavity. The bony defect may continue cephalad along the nasomaxillary process to the level of the lacrimal bone and orbital rim and floor, resulting in vertical orbital dystopia. In the most severe cases, absence of bone along this axis results in a confluence of the orbital, maxillary, nasal, and oral cavities (Fig. 6‑5).
Number 4 Cleft
This cleft is also oblique in nature, but unlike the numbers 0 to 3 clefts, the lip defect occurs laterally, between the philtral ridge and the oral commissure. In addition, the defect moves onto the cheek and largely spares the subunits of the nose, despite the shortened alar–ocular distance. The medial canthus is largely spared, as the cleft transitions onto the lower eyelid, lateral to the punctum. The lacrimal drainage structures are therefore intact but often dysfunctional. The globe can show anophthalmia, microphthalmia, or normal anatomy. The alveolar defect also occurs between the lateral incisor and cuspid, but the pyriform is spared. The bony cleft extends through the maxilla medial to the infraorbital foramen and then into the orbit. Herniation of orbital contents yields a hypoplastic and dystopic orbit. In the complete form of the number 4 cleft, the oral, maxillary, orbital cavities are confluent, for which reason this cleft has been referred to as the oro-ocular oblique cleft (Fig. 6‑6).
Number 5 Cleft
The number 5 cleft is the rarest of the oblique facial clefts and is rarely seen in isolation. Its clinical characteristics are similar to those of the number 4 cleft. The maxillary defect occurs lateral to the infraorbital foramen and then across the rim and onto the floor of the lateral orbit.
Number 6 Cleft
The number 6, 7, and 8 rare craniofacial clefts often coincide as Treacher Collins syndrome (TCS), and the isolated number 7 cleft underlies the findings associated with craniofacial microsomia. Both of these clinical entities are described in detail elsewhere in this text and will therefore not be discussed in depth here. However, there are relevant aspects of these individual clefts that merit independent discussion.
In contrast to the syndromic presentation, which is characterized by absence of the zygoma and its arch, the isolated number 6 cleft is defined by a present but hypoplastic zygomatic body and an intact zygomatic arch. Soft tissue manifestations are similar to those seen in patients with TCS but are milder in nature (Fig. 6‑7).
Number 7 Cleft
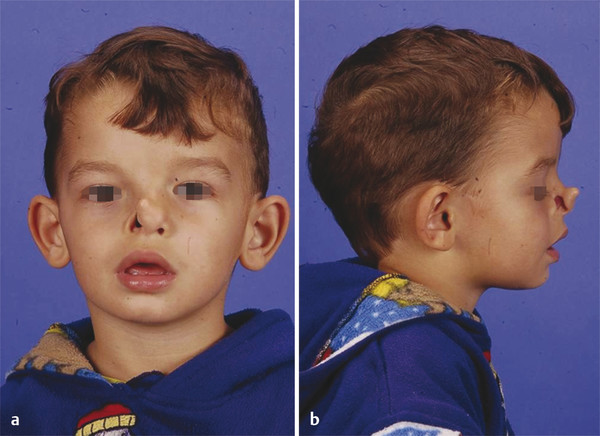
In addition to its role in TCS, the number 7 cleft is the most common isolated rare craniofacial cleft. The numerous ways in which it has been historically referred to indicate the variability in its clinical manifestation: hemifacial microsomia, craniofacial microsomia, microtia, otomandibular dysostosis, first and second branchial arch syndrome, and oromandibuloauricular syndrome. Macrostomia is the hallmark feature of the cleft, but soft tissue involvement can be diffuse, ranging from small preauricular skin tags to significant deficiency and distortion of the auricle, cheek, tongue, soft palate, parotid gland, and facial musculature. The cardinal bony defects are centered on the zygomaticotemporal suture, with resultant hypoplasia or absence of the zygoma or mandible (Fig. 6‑8).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


