The connective tissue (collagen vascular, rheumatic) disorders represent a group of diseases characterized by inflammatory changes of the connective tissue in various parts of the body. Among these, juvenile idiopathic arthritis (JIA), lupus erythematosus, juvenile dermatomyositis (JDM), systemic and localized forms of scleroderma, eosinophilic fasciitis, Sjögren syndrome, mixed connective tissue disease (MCTD), and antiphospholipid antibody syndrome (APS) exhibit specific cutaneous findings.
Juvenile Idiopathic Arthritis
Juvenile idiopathic arthritis (JIA) is a group of seven arthritides of children under the age of 16 years that last longer than 6 weeks and are of unknown cause. JIA encompasses the former term juvenile rheumatoid arthritis, as well as other arthritides in children. These include systemic arthritis (Still disease), oligoarthritis, rheumatoid factor (RF)-negative polyarthritis, RF-positive polyarticular arthritis, psoriatic arthritis, and enthesitis-related arthritis ( Box 22-1 ). Overall, juvenile arthritis has a prevalence of up to 1 : 1000 children under the age of 16 years. Approximately 6% of these children have systemic-onset disease; 13%, RF-negative polyarticular arthritis; 2%, RF-positive polyarticular arthritis; 52%, oligoarthritis; 12%, enthesitis-related arthritis; 8%, psoriatic arthritis (see Chapter 4 ); and 8%, undifferentiated arthritis. However, subtype distribution varies among countries; for example, although oligoarthritis is most common in Europe, polyarthritis is more common in New Zealand and India. RF, usually an immunoglobulin (Ig) M antibody against IgG, rarely occurs in children, especially children under 7 years of age, and is associated with greater synovial erosion with a poor prognosis. Antinuclear antibody (ANA) levels (which are positive in 10% of normal children, usually at a level of 1 : 40 to 1 : 80) are most commonly positive in children with oligoarticular disease (40% to 80%), especially in girls and with uveitis. The different subtypes of JIA are associated with human leukocyte antigen (HLA) types. Oligoarthritis is associated with HLADR8, HLADR6, and HLADR5, with chronic uveitis in oligoarthritis linked to HLADR5. RF-positive polyarticular JIA has been associated with HLADR4, whereas RF-negative polyarthritis has been linked to HLADR8, HLADPw3, and HLADQw4. Systemic-onset disease has been associated with HLADR4, HLADR5, and HLADR8, and the enthesitis-related arthritis (juvenile spondylitis) with HLAB27 and a striking familial occurrence. Patients with JIA and coexisting atopy have been shown to have more active disease and worse outcomes, possibly because of ongoing immune dysregulation.
Systemic-Onset Arthritis
Definition: Arthritis with or preceded by daily fever of at least 2 weeks’ duration plus at least one of the following:
Evanescent, nonfixed erythematous eruption
Generalized lymphadenopathy
Serositis
Hepatosplenomegaly
Oligoarthritis
Definition: Arthritis that affects one to four joints during the first 6 months of disease. Two subtypes occur: 1) persistent oligoarthritis that never affects more than four joints; and 2) extended oligoarthritis that affects a total of five or more joints after the first 6 months of the disease
Exclusions:
Family history of confirmed psoriasis in at least one first- or second-degree relative
Family history of confirmed HLA B-27-associated disease in at least one first- or second-degree relative
HLA-B27 positivity in a boy with the onset of arthritis after 8 years of age
Positive RF testing
Presence of systemic-onset arthritis as defined
Polyarthritis (RF-negative)
Definition: Arthritis that affects five or more joints during the first 6 months with RF-negative testing at least twice and at least 3 months apart. Systemic-onset JIA must be excluded
Polyarthritis (RF-positive)
Definition: Arthritis that affects five or more joints during the first 6 months of disease, associated with a positive RF test twice and at least 3 months apart. Systemic-onset JIA must be excluded
Psoriatic Arthritis
Definition: Must have arthritis and psoriasis, or arthritis and at least two of the following:
Dactylitis
Nail pitting or onycholysis
Family history of confirmed psoriasis in at least one first-degree relative
Enthesitis-Related Arthritis
Definition: Arthritis and enthesitis, or arthritis or enthesitis and at least two of the following:
Sacroiliac joint tenderness and/or inflammatory spinal pain
HLA-27 positivity
Family history of at least one first- or second-degree relative with confirmed HLA-27–associated disease
Anterior uveitis that is usually symptomatic (pain, redness, photophobia)
Onset of arthritis in a boy after the age of 8 years
Systemic-onset JIA and confirmed psoriasis in at least one first- or second-degree relative must be excluded
HLA, Human leukocyte antigen; RF, rheumatoid factor; JIA, juvenile idiopathic arthritis.
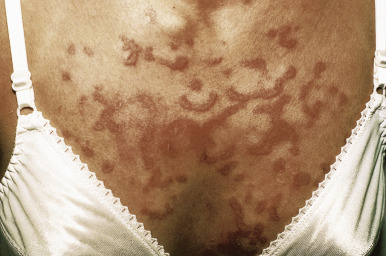
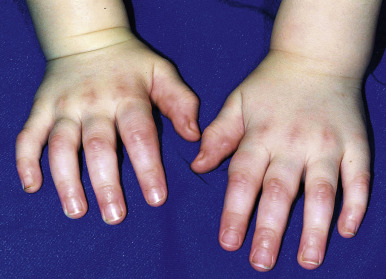
Of the various forms of JIA other than psoriatic arthritis, systemic-onset disease is the only one with known cutaneous findings. Its underlying pathomechanism involving dysfunction in the innate immune system, its clinical features, and its poor response to therapies that ameliorate other forms of JIA have led several investigators to classify systemic JIA as an auto-inflammatory, rather than autoimmune, disease (see Chapter 25 ). Systemic-onset disease affects boys and girls equally. The eruption of systemic-onset JIA is usually intermittent, occurring most often when patients have their characteristic daily or twice daily spiking fevers. The fevers generally reach 39° C or higher, and the children appear acutely ill with exacerbation of joint pain when fevers occur. The characteristic flat to slightly elevated macules or occasionally papules may resemble urticaria but are not pruritic. They typically measure 2 mm to 6 mm in diameter. Lesions vary from salmon-pink to red and display a characteristic slightly irregular or serpiginous margin. The blanching macules are often evanescent and commonly subside during periods of remission and a few hours after defervescence. Individual lesions may coalesce to form large plaques 8 to 9 cm in diameter. The eruption occurs predominantly on the trunk but often affects the extremities and occasionally the face ( Fig. 22-1 ); it is accentuated by local heat or trauma. Seen in 25% to 50% of patients, it may precede fevers or visceral involvement by up to 3 years. In some patients the rash occurs during a period of only 1 week, in others for a year or more. Persistent pruritic papules and plaques can occur in children with systemic JIA (and more often in adults). Biopsy helps confirm the diagnosis and shows dyskeratosis of the upper epidermis and sparse inflammation.
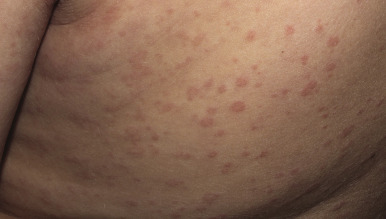
Many patients have extraarticular features, particularly hepatosplenomegaly, pleuritis, pericarditis, and lymphadenopathy, which may predate the joint manifestations by weeks to, rarely, years. Approximately 50% of affected individuals have a mild oligoarticular course with a good prognosis for remission; of the remaining 50%, approximately half will develop severe, recalcitrant, and destructive progressive polyarticular arthritis. Other complications of systemic-onset JIA include secondary amyloidosis, secondary infection, osteoporosis, and growth retardation.
Oligoarticular JIA occurs more often in girls than in boys and peaks between the ages of 2 and 4 years. Children with oligoarticular JIA often limp and show joints that are warm and effused, but not red and hot. In about 50% of cases, the onset is monoarticular with the knee, ankle, or elbow most commonly affected; small joints are usually spared. The acute onset of painful monoarthritis with refusal to bear weight is unlikely to be JIA, and infectious, traumatic, and malignant causes of joint pain should be considered. Approximately 20% to 30% of children with JIA have uveitis, which occurs most commonly in ANA-positive girls with early onset oligoarthritis. The classic picture is a chronic bilateral anterior uveitis, which is usually asymptomatic until significant, and sometimes irreversible damage to intraocular structures occurs. Patients with oligoarticular JIA and uveitis have worse ocular outcomes relative to patients with other causes of uveitis, necessitating regular screening examinations. Treatment of the uveitis includes topical corticosteroids, mydriatics, systemic immunosuppressive agents, and surgical management of complications.
Polyarthritis usually has an insidious onset, although it may be acute. It can involve both large and small joints (particularly of the hands and feet), and commonly involves the temporomandibular joints and cervical spine. Lymphadenopathy, hepatomegaly, and fevers may occur, although the fevers do not show the daily (quotidian) spikes seen in systemic JIA.
Children with enthesitis-related arthritis have inflamed tendons in addition to arthritis of one or more joints. Most affected individuals are boys over the age of 10 years. Although children rarely show typical ankylosing spondylitis at onset, spondyloarthropathy not uncommonly develops by adulthood. Arthritis associated with inflammatory bowel disease is a subset of this type. Acute anterior uveitis can occur in up to 27% of children and tends to present as an acutely painful, red, photophobic eye in contrast to the asymptomatic uveitis seen in oligoarticular JIA.
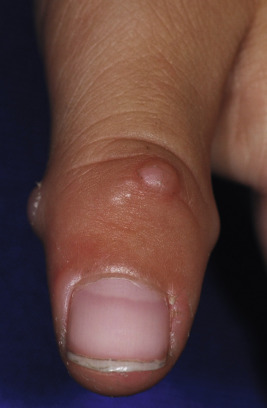
Although more characteristic of SLE, scleroderma, and dermatomyositis, patients with JIA may also demonstrate cuticular telangiectasias. Subcutaneous nodules rarely are seen in children with JIA and occur most commonly in patients with RF-positive polyarthritis, particularly if the disease is recalcitrant to therapy. Barely palpable to several centimeters in size, they may be the first presenting sign of JIA. Their most common location is near the olecranon process on the ulnar border of the forearm. Less commonly they may occur on the dorsal aspect of the hands, on the knees and ears, and over pressure areas such as the scapulae, sacrum, buttocks, and heels. In the areas of fingers and toes, subcutaneous nodules are only a few millimeters in size. Subcutaneous nodules are firm and nontender and may be attached to the periarticular capsules of the fingers. They can be easily confused with the subcutaneous form of granuloma annulare (see Chapter 9 ). Rheumatoid nodules and JIA must also be distinguished from fibroblastic rheumatism, a rare disorder characterized by the sudden but sometimes progressive onset of cutaneous nodules ( Fig. 22-2 ), flexion contractures, and polyarthritis. Some affected individuals respond to methotrexate or interferon (IFN)-α. Methotrexate and tumor necrosis factor (TNF) inhibitor treatment have been linked to induction of subcutaneous nodules, most often in rheumatoid arthritis, but this nodulosis has only been described in adults.
Traditional nonsteroidal anti-inflammatory drugs (NSAIDs) continue to be the first-line therapy in all but those with systemic JIA, in addition to physical therapy and psychosocial support. NSAIDs that preferentially inhibit the cyclooxygenase-2 (COX-2) enzymes have fewer gastrointestinal (GI) adverse effects and may be preferred in some patients. Pseudoporphyria, presenting with tense blisters in photodistributed sites, has most commonly been associated with the use of naproxen sodium but has been reported after administration of other NSAIDs as well, including COX-2 inhibitors (see Chapter 19 ).
Intraarticular injections of corticosteroids are used for children who do not respond to NSAID therapy or as initial therapy for oligoarthritis. Methotrexate is usually the second line of treatment for persistent, active arthritis but is best initiated early in the disease course. Patients with oligoarticular arthritis tend to respond most favorably. Blockade of TNF with biologics (etanercept, infliximab, adalimumab) in combination with methotrexate, is particularly effective for children with polyarticular arthritis. Sulfasalazine has largely been used for enthesitis-related arthritis.
Systemic-onset JIA responds poorly to NSAIDs and TNF-α blockers (30% response) but will typically respond to corticosteroids and often to anakinra or canakinumab (anti-interleukin [IL]-1 receptor antagonists), tocilizumab (humanized anti-IL-6 receptor monoclonal antibody), abatacept (T cell-activating agent/IgG1 fusion protein), or methotrexate. Moderate or high dosages of systemic corticosteroids, including pulse intravenous steroids to avoid long-term high-dose treatment, are occasionally used for short periods while awaiting the effects of other medications. As a chronic inflammatory disorder and in relation to usage of systemic corticosteroids, JIA is associated with an increased risk of osteoporosis, which can be countered by treatment with calcium and vitamin D supplementation, bisphosphonates, and calcitonin.
Macrophage Activation Syndrome
Macrophage activation syndrome (MAS), a severe, life-threatening complication of systemic-onset JIA and other connective tissue diseases, is associated with hemophagocytosis of activated macrophages/histiocytes and multiorgan dysfunction. When observed in the absence of underlying connective tissue disease, this diagnosis is termed hemophagocytic lymphohistiocytosis ( HLH ) and can be primary (when familial) or secondary (typically in the setting of infection or malignancy) (see Chapter 10 ). The pathophysiology of both MAS and HLH are speculated to involve inability of natural killer (NK) or CD8 T cells to lyse infected antigen-presenting cells, leading to ongoing infection and resultant “cytokine storm.” Patients show the sudden onset of fever, hemophagocytosis with hepatic dysfunction, disseminated intravascular coagulation, and sometimes encephalopathy, respiratory distress, and renal failure. The disease may present as pancytopenia with a falling erythrocyte sedimentation rate (ESR), in contrast to the increased ESR associated with flaring JIA. Formal diagnostic criteria for MAS are currently lacking, but a recent international consensus survey of pediatric rheumatologists revealed the 10 most useful diagnostic features: decreasing platelet count, hyperferritinemia, evidence of macrophage hemophagocytosis in the bone marrow, increased liver enzymes, decreasing leukocyte count, persistent continuous fever greater than 38° C, falling ESR, hypofibrinogenemia, and hypertriglyceridemia. Patients with MAS and systemic JIA have been shown to harbor defects in NK-cell function and in genes that encode proteins crucial to lymphocytic cytolytic activity, including Munc 13-4 and perforin, which are also implicated in primary familial hemophagocytic lymphohistocytosis. Early recognition of MAS and treatment with high-dose corticosteroids with or without cyclosporine, etoposide, and/or etanercept may be life saving.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease characterized by autoantibody accumulation with immune complex deposition leading to end-organ damage. It often involves the skin but may affect almost any organ system. Autoantibodies are the hallmark of SLE, and activated B cells, T helper cells, and the cytokines that modulate inflammatory responses are thought to be the most appropriate targets for therapy. Both genetic and environmental factors are thought to play roles in its pathogenesis. Reports of familial cases and a high concordance for clinical disease in monozygotic, but not dizygotic, twins support a strong genetic contribution to the development of SLE. SLE has been linked to several genetic susceptibility loci that encode protein products involved in innate and adaptive immunity, particularly in pathways of apoptosis and immune complex clearance, activation of toll-like receptors (TLRs), and signaling of type I IFN and nuclear factor (NF)-κB. The availability of whole exome sequencing has led to the discovery of pathogenic mutations in autosomal recessive juvenile-onset SLE ( PRKCD , encoding protein kinase Cδ), complement deficiency leading to SLE, and autosomal recessive hypocomplementemic urticarial vasculitis ( DNASE1L3 ). Although most individuals with SLE do not have monogenic disease, these discoveries may increase understanding of the pathogenesis and lead to new ideas for therapy.
Environmental triggers, including ultraviolet (UV) light, medications, pesticides, heavy metals, tobacco, and infections, have all been linked to the development of SLE. In particular, exposure to UV light is a trigger, not only for cutaneous manifestations, but also for flares of systemic disease.
Approximately 15% to 20% of all cases of SLE occur within the first 2 decades of life. Of these, 60% occur between the ages of 11 and 15 years, 35% between 5 and 10 years, and 5% in children younger than 5 years of age. Other than neonatal lupus erythematosus (NLE), the disorder is rarely seen before the age of 3 years. Disease onset in the first decade of life is associated with a female to male ratio of 4 : 3, which increases to female predominance in the second decade (4 : 1), compared with a ratio of 9 : 1 in patients with adult-onset SLE. The overall prevalence is 5 to 10 out of 100,000 children, and both increased prevalence and severity are seen in African-American and Hispanic children, especially because of the increased prevalence of nephritis. In comparison with SLE in adults, affected children have more severe disease in general, with an increased incidence of renal, neurologic and hematologic involvement at the time of diagnosis. In one study, 77% of pediatric patients required moderate to high dosages of corticosteroids, compared with 16% of adult patients.
About 80% of patients with SLE have cutaneous involvement at some time, often as the presenting sign. Diagnosis of SLE requires Systemic Lupus International Collaborating Clinics (SLICC) criteria that were established in 2012 ( Table 22-1 ) and are more accurate at classification than the previous criteria established by the American College of Rheumatology (ACR) in 1982. The 1982 criteria included four mucocutaneous features: the malar eruption, discoid eruption, photosensitivity, and oral ulcerations. In 2012, the dermatologic criteria for SLE were broadened to include the variety of subtypes of cutaneous lupus under either acute or chronic cutaneous lupus criteria. In addition, to prevent duplication of highly correlated terms related to cutaneous lupus, photosensitivity was replaced with diffuse, nonscarring alopecia.
| Criterion * | Comments | |
|---|---|---|
| CLINICAL CRITERIA | ||
| 1 | Acute cutaneous lupus | Lupus malar rash (fixed erythema, flat or raised, over malar eminences, usually sparing nasolabial folds), bullous lupus, photosensitive lupus rash (not dermatomyositis), maculopapular lupus rash, or toxic epidermal necrolysis variant Subacute cutaneous lupus (nonindurated annular polycyclic or psoriasiform lesions that resolve without scarring) |
| 2 | Chronic cutaneous lupus | Classic discoid eruption (erythematous plaques with adherent keratotic scaling and follicular plugging, often atrophic scarring and dyspigmentation if older lesion; localized if only above the neck, otherwise generalized), lupus panniculitis (profundus), hypertrophic (verrucous) lupus, lupus tumidus, chilblains lupus, discoid lupus/lichen planus overlap, mucosal lupus |
| 3 | Oral ulcers | Palate, buccal, tongue, or nasal mucosa (no other cause identified) |
| 4 | Alopecia, nonscarring | Diffuse thinning or hair fragility in the absence of other causes (such as alopecia areata, iron deficiency, drugs, or androgenetic alopecia) |
| 5 | Synovitis | Must involve two or more joints with swelling or effusion or tenderness and at least 30 minutes of morning stiffness |
| 6 | Serositis | Pleuritis, pleural effusions or pleural rub; pericardial pain, effusion, rub or pericarditis without other cause |
| 7 | Renal | Persistent proteinuria >0.5 g/day or red blood cell casts |
| 8 | Neurologic | Seizures, psychosis, myelitis, mononeuritis multiplex, peripheral or cranial neuropathy, acute confusional state (all without other known cause) |
| 9 | Hemolytic anemia | |
| 10 | Leukopenia | <4000/mm 3 at least once or lymphopenia of <1000/mm 3 at least once without other cause |
| 11 | Thrombocytopenia | <100,000/ mm 3 at least once without other cause |
| IMMUNOLOGIC CRITERIA | ||
| 1 | Positive ANA | Above laboratory reference range |
| 2 | Anti-ds DNA antibody | Above laboratory reference range (>2-fold if by ELISA) |
| 3 | Anti-Sm antibody + | |
| 4 | Antiphospholipid antibody + | + Lupus coagulant or anti-β2-glycoprotein I, medium- or high-titer anticardiolipin antibody, or false positive |
| 5 | Low complement | C3, C4, CH50 |
| 6 | Direct Coombs test | In the absence of hemolytic anemia |
* At least four criteria are required, including at least one clinical and one immunologic criterion. Biopsy-proven lupus nephritis plus a positive ANA or anti-ds DNA is also sufficient.
Cutaneous Lupus Erythematosus
Cutaneous lupus erythematosus (CLE) may occur as an isolated finding or in the setting of underlying SLE. Features of CLE may be classified as lupus-specific or nonspecific, based on clinical and histopathologic characteristics. Lupus-specific subsets of cutaneous disease characteristically demonstrate lupus-specific features on histopathology, including interface dermatitis and mucin deposition. These include acute CLE (ACLE), subacute CLE (SCLE), intermittent CLE (ICLE), and chronic cutaneous lupus erythematosus (CCLE). Lupus-nonspecific cutaneous disease includes a variety of reactive phenomena, which are not pathognomonic or specific for SLE but typically occur in active disease.
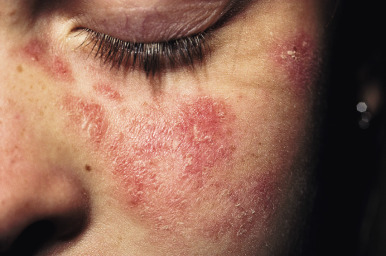
ACLE may present with a malar or butterfly rash or a more generalized morbilliform photodistributed eruption. In particular, the malar rash, while seen in approximately 60% of patients with SLE, is not specific, but when present is often a sign of underlying systemic disease. This erythematous, mildly scaling eruption ( Fig. 22-3 ) often appears over the cheeks and bridge of the nose, resembling the shape of a butterfly. It can be confused with the malar erythema of seborrheic dermatitis, erythematotelangiectatic acne rosacea, parvovirus infection, or cutaneous lichenoid graft-versus-host disease ; seborrheic dermatitis typically involves the nasolabial fold and shows scaling as well. Brightly erythematous papules and plaques can also be seen on the photo-exposed extremities and V-shaped region of the neck. When this eruption presents on the dorsal aspect of the hands in SLE, the areas overlying joints are typically spared ( Fig. 22-4 ), in contrast to JDM.
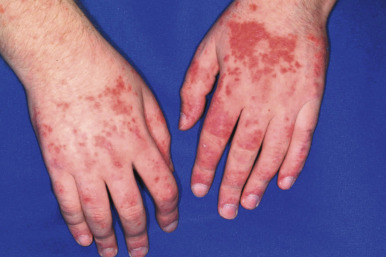
SCLE, which represents 10% to 15% of all cases of CLE, only occasionally occurs in pediatric patients. The cutaneous lesions of SCLE arise quite suddenly, mainly on the upper trunk, extensor surfaces of the arms, and dorsal aspect of the hands and fingers. The lesions may be annular and may enlarge and merge into psoriasiform or polycyclic lesions with thin and easily detached scales ( Fig. 22-5 ). UV light is usually associated with exacerbations, but lesions tend to be nonscarring. Although arthritis and fatigue are common, patients usually only have mild systemic manifestations. The majority of patients with SCLE have anti-Ro (SS-A) antibodies and many have anti-La antibodies. Nonbullous annular lesions that clinically and histologically resemble Sweet syndrome (see Chapter 20 ) can also been seen as a manifestation of SLE; biopsy shows unique histologic features called the histiocytoid form of neutrophilic dermatosis. Rowell syndrome was originally described as an erythema multiforme-like eruption in association with lupus (classically CLE), anti-La antibodies, and speckled ANAs; although controversy exists, most of these cases are probably SCLE or the concurrence of lupus and erythema multiforme. SCLE is treated by strict sun protection, topical anti-inflammatory agents, antimalarials, and occasionally systemic corticosteroids.
Tumid lupus (lupus erythematosus tumidus) is a form of ICLE. Red to purplish urticarial plaques, which are relatively fixed in shape and do not undergo atrophy or scaling, usually occur on the face or other exposed areas of the body as a sign of photosensitivity. The progression rate of lupus erythematous tumidus to SLE is unknown.
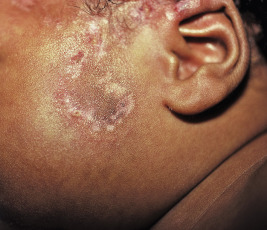
CCLE is an umbrella term used to describe lesions of discoid lupus erythematosus (DLE), lupus panniculitis, and chilblain lupus. Individual lesions of DLE are well-circumscribed, elevated, indurated red to purplish plaques with adherent scale ( Figs. 22-6 and 22-7 ) and fine telangiectasia. When untreated or as a sequela, discoid lesions may develop persistent areas of hyperpigmentation or hypopigmentation with atrophy. Discoid lesions can be asymmetric and are often exacerbated by exposure to UV light. DLE is most commonly found on the face, often with lesions on the ear helices and conchal bowls. Although the face is often the sole site of DLE without SLE, the scalp, arms, legs, hands, fingers, back, chest, abdomen, and even mucosal areas ( Fig. 22-8 ) may also be involved. At times the openings of hair follicles are dilated and plugged by an overlying scale. If the scale is thick enough, it can be lifted off in one piece. The undersurface then reveals follicular projections that resemble carpet tacks, a characteristic sign of LE. Discoid lesions have been described in a linear orientation that follows the lines of Blaschko.
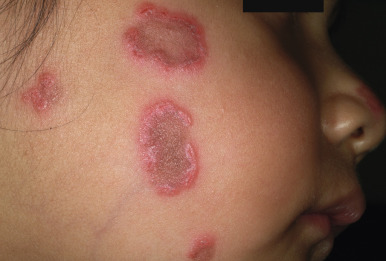
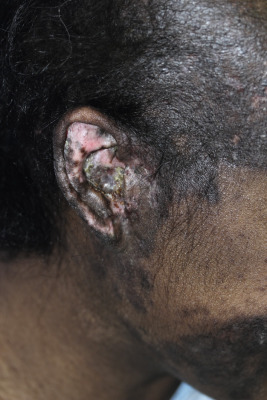
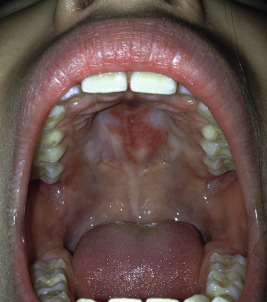
In childhood, DLE is rare, and less than 3% of patients with DLE develop disease before the age of 10 years. DLE may present as an isolated finding or, more commonly, as a manifestation of underlying SLE. Some patients develop full criteria for SLE over time, mandating serial clinical and serologic evaluations for systemic disease. In adults, the rate of progression of DLE to SLE is reported to range from 0 to 28%, with an interval between onset of DLE and SLE ranging from months to decades. Some studies in both adults and children have demonstrated that patients with generalized lesions (above/below the neck) are at higher risk for SLE. In addition, adults with rising ANA titers, elevated ESR levels, anemia, leukopenia, arthralgias, nail changes, and evidence of nephropathy may predict progression to systemic disease. Small studies in children have demonstrated an early rate of progression to SLE approaching 25% to 30% during a variable follow-up duration of months to years. However, children with DLE who develop SLE appear to follow a benign course of systemic disease, with 89% of patients in one study meeting SLE classification criteria with mucocutaneous limited manifestations and no evidence of end-organ damage over 5 years of follow-up study.
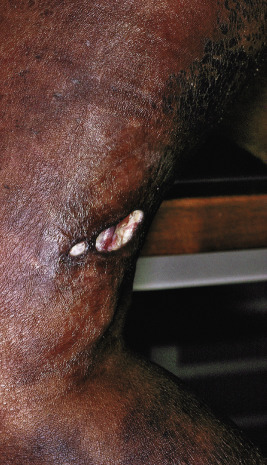
Intense inflammation of the fat (lupus panniculitis, lupus profundus) occurs in approximately 2% of patients with CLE, and may be associated with DLE, SLE, or, not uncommonly, as an isolated disorder. Lesions are seen as firm, often asymptomatic, sharply defined, rubbery dermal and subcutaneous plaques or nodules with a predilection for the scalp, forehead, cheeks, upper arms, breasts, thighs, and buttocks. Although the skin overlying the lesions usually appears normal, it often ulcerates ( Fig. 22-9 ) or becomes depressed. Biopsy shows a perivascular lymphocytic, plasma cell, and histiocytic infiltration in the deep dermis and subcutaneous fat without accompanying fat necrosis. The panniculitis may be lobular alone or involve both septae and lobules; most specimens show mucin deposition. Direct immunofluorescence often reveals Igs or C3 deposits at the basement membrane. Approximately 70% of patients with lupus panniculitis also have typical lesions of DLE, and 50% eventually develop systemic involvement or symptoms of SLE such as fever, arthralgia, and lymphadenopathy. Lesions of lupus profundus may persist for years and leave significant disfigurement. Although erythema nodosum (see Chapter 20 ) may occur in a patient with SLE, lupus panniculitis can often be distinguished by its lack of tenderness, greater chronicity, and lack of predilection for the lower legs. Lupus panniculitis must also be distinguished from subcutaneous lymphoma by histologic features and T-cell rearrangement studies. Inflammatory panniculitis followed by lipoatrophy, likely autoimmune in mechanism but not associated with SLE or dermatomyositis, can involve localized areas, such as the ankles or abdomen. Lipoatrophic panniculitis can also be seen in type 1 diabetes, Hashimoto thyroiditis, and JIA, which may manifest years after the onset of the panniculitis. The tender erythematous nodules demonstrate lobular panniculitis on histopathology. With clearance, these nodules leave lipoatrophy that may progress to partial or even generalized lipodystrophy. Administration of systemic corticosteroids and/or steroid-sparing agents such as methotrexate can stop the development of nodules. Lipodystrophy and tender erythematous nodules are also a feature of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE syndrome), a proteasome auto-inflammatory disorder (see Chapters 20 and 25 ).
Nonspecific Manifestations of SLE
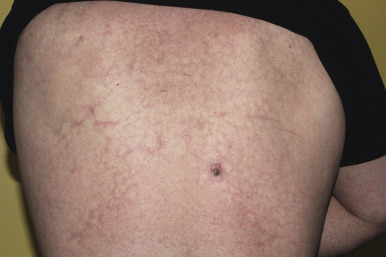
Nonspecific manifestations of SLE are distinguished by the absence of lupus-specific changes on histopathology. These include a variety of reactive and inflammatory eruptions and are typically observed in active SLE. Livedo reticularis ( Fig. 22-10 , see also Fig. 12-81 ), blotchy bluish-red discoloration of the skin due to vasospasm, is seen in several collagen vascular disorders, among them SLE, drug-induced SLE, APS, and scleroderma. Aggravated by exposure to cold, the reticulated erythema tends to be persistent and is most commonly found on the lower extremities. Raynaud phenomenon, seen in 10% to 30% of patients with SLE, may precede the onset of SLE by months or years. This phenomenon is described in more detail in the section on systemic sclerosis (SSc). Pernio (chilblains) are tender cyanotic to reddish-blue nodular swellings on the fingers or toes (see Chapter 20 ). Chilblain lupus manifests as cold-induced, reddish-blue, inflammatory acral lesions that may ulcerate (see Chapter 20 ). Although sporadic chilblain lupus has been described in adolescents, most of the affected younger children have a familial form of chilblains (see Chapter 20 ). Chilblains must be distinguished from stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI syndrome), a recently described auto-inflammatory disorder with acral vasculopathy that can be exacerbated by cold (see Chapter 25 , Fig. 25-26 ).
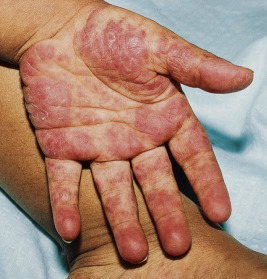
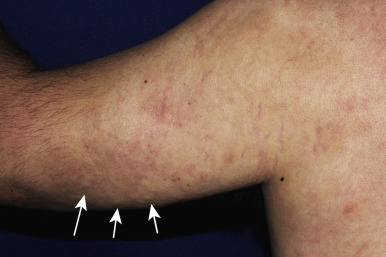
Chronic urticaria-like lesions (urticarial vasculitis) in children with SLE may be distinguished from classic urticaria by their tendency to persist longer than 24 hours and their lesser tendency to be pruritic (see Chapter 21 , Fig. 21-14 ). A manifestation of immune complex deposition, urticarial vasculitis generally occurs in patients demonstrating clinical or serologic evidence of systemic disease activity, especially hypocomplementemia. Biopsy confirms the presence of leukocytoclastic vasculitis. Leukocytoclastic vasculitis can also be seen in biopsy specimens from purpuric lesions in patients with SLE. These purpuric lesions may ulcerate, leaving significant scarring. Erythema and papular telangiectasia of the palms and fingers ( Fig. 22-11 ), linear telangiectasia of the cuticles and periungual skin (with or without thromboses), and periorbital or hand and feet edema, are other nonspecific cutaneous features of SLE.
Although bullous or crusted lesions of SLE may simply reflect fragility of skin because of liquefaction degeneration of basal keratinocytes, a distinctive complication of SLE resembles bullous or epidermolysis bullosa acquisita (EBA) and has been called bullous (or vesiculobullous ) SLE ( BSLE ; see Chapter 13 , Fig. 13-32 ). Indirect immunofluorescence on salt-split skin shows Ig and C3 deposits on the dermal side, and immunoblotting shows autoantibodies directed against type VII collagen, as in patients with EBA (see Chapter 13 ). Subtle variations in IgG subtypes between EBA and BSLE have been shown by IgG subtyping. Other forms of immunobullous disorder, such as linear IgA disease, have also been described in association with SLE, emphasizing the need for immunofluorescence testing.
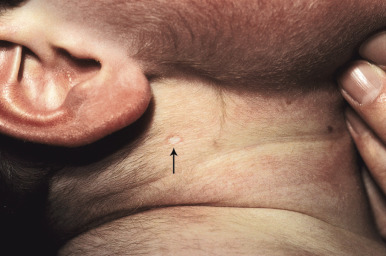
Mucin deposition is a typical characteristic seen on skin biopsy sections of individuals with SLE. However, skin-colored, sometimes pruritic papules and nodules involving primarily the trunk, upper extremities, and occasionally the face are a clinical feature of SLE that reflects large amounts of mucin in the mid-dermis between collagen bundles ( Fig. 22-12 ). Papulonodular (dermal) mucinosis of SLE may occur any time during the course, including before onset of other features, and has a course that is independent of the course of the SLE. Mucinosis in children is also a histologic feature of self-healing juvenile cutaneous mucinosis (SHJCM), an unusual disorder characterized by the acute onset of multiple skin-colored to dusky erythematous asymptomatic papules and nodules. Lesions mainly occur on the head and periarticular areas, and can be a few centimeters in diameter ( Fig. 22-13 ), in contrast to the more diffuse and, if discrete, papular lesions of the papulonodular mucinosis of SLE. Fever, muscle tenderness, arthralgias, and weakness may be associated. Lesions spontaneously resolve during the subsequent months to a few years. Biopsy samples can be erroneously misdiagnosed as panniculitis or proliferative fasciitis, but mucin stains allow the characteristic deposition of large amounts of mucin to be identified so that the family can be reassured.
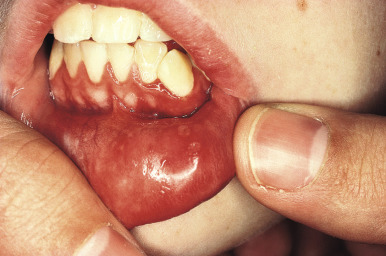
Mucosal membrane lesions (gingivitis, mucosal hemorrhage, erosions, and small ulcerations) are seen in 3% of patients with CLE and in up to 30% of pediatric patients with the systemic form of the disorder. A silvery whitening of the vermilion border of the lips is highly characteristic, and the lips may show slight thickening, roughness, and redness, with or without superficial ulceration and crusting ( Fig. 22-14 ). The gingival, buccal, and nasal mucosae may also appear red, edematous, friable, and eroded, or may exhibit silvery white changes.
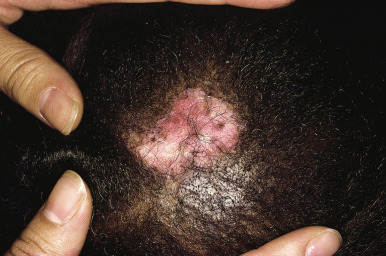
Several forms of alopecia may occur in patients with SLE (overall in about 30% of patients). First, discoid lupus may present with scarring alopecia associated with multiple well-demarcated patches ( Fig. 22-15 ) that exhibit erythema, scaling, telangiectasia, atrophy, and plugging (the classic changes of DLE). Second, patients with SLE may develop hair fragility, especially with acute exacerbations, that leads to hair breakage several millimeters from the roots, resulting in a receding hairline with a short, unruly, broken hairs (“lupus hair”), especially at the temple and forehead. Telogen effluvium may also occur in patients with SLE. Finally, there is an increased incidence of alopecia areata in patients with SLE, and these patients tend to have classic scalp dermoscopic findings including exclamation-mark hairs, black dots, broken hair, and yellow dots (see Chapter 7 ).
The most common symptoms of SLE at presentation are constitutional (malaise, fever, and weight loss). The arthritis, which occurs in up to 79% of pediatric patients, is a symmetric polyarthritis involving both small and large joints. It is commonly quite painful (in contrast to the arthritis of JIA), often out of proportion to the clinical findings, and tends to be nondeforming. Approximately 55% of pediatric patients develop renal involvement, most often during the first year of the SLE. Four types of nephritis have been described based on renal biopsy pattern: mesangial lupus nephritis, focal proliferative, diffuse proliferative, and membranous glomerulonephritis. Diffuse proliferative glomerulonephritis occurs most commonly (40% to 50%) but carries the worst prognosis, leading to end-stage renal disease in approximately 10% to 15% of patients with lupus nephritis. The presence of anti-double-stranded (anti-ds) deoxyribonucleic acid (DNA) antibodies and hypocomplementemia is commonly associated with severe renal damage. Pulmonary involvement, particularly pleuritis, occurs in up to 30% of patients. Cardiac manifestations, including pericarditis, myocarditis, vasculitis affecting coronary arteries, and Libman-Sachs endocarditis, occur in approximately 15% of patients. A recently recognized issue, however, is the long-term risk of accelerated atherosclerosis ; as a result, the overall survival for SLE has improved for all causes except cardiovascular disease. Individuals with childhood-onset SLE can develop myocardial ischemia as early as 20 to 30 years of age. Neuropsychiatric SLE (central nervous system [CNS] involvement) most commonly features headache, seizures, alterations in mental status, psychosis, and peripheral neuropathy, and is seen in 20% to 30% of affected children ; most show evidence of neuropsychiatric involvement during the first year of the disease, but cognitive dysfunction occurs in almost 25%. Neuroimaging studies may show structural abnormalities, such as infarction; the addition of single-photon emission computed tomography (SPECT) scans may show evidence of cerebral vasoconstriction and vasculitis as perfusion abnormalities and allow a diagnosis of lupus psychosis. Lymphadenopathy, although it occurs more commonly in adults, is seen in up to 40% of children with SLE. Ocular findings are seen in 25% of patients (cotton-wool patches, optic neuropathy, perivasculitis, and edema of the optic disk).
The diagnosis of SLE is chiefly clinical and is based on the presence of cutaneous lesions, systemic manifestations, and confirmatory laboratory tests (see Table 22-1 ). Biopsy of cutaneous lesions will confirm the diagnosis and shows: 1) hyperkeratosis with keratotic plugging; 2) epidermal atrophy; 3) liquefactive degeneration of basal cells; and 4) a patchy, chiefly lymphoid cell infiltrate, especially around appendages and vessels. Biopsy of lesional skin in DLE or SLE usually shows the deposition of immunoreactants at the epidermal–dermal border (the lupus band test). Sun-exposed areas should be avoided for this biopsy because of the risk of a false-positive lupus band test at sun-exposed sites.
Serologic examination is the most important evaluation for children thought to have SLE, and ANA is the most valuable screening test. The serologic profiles in children with SLE are similar to those of adults, although some authors have reported a slightly increased incidence of positive antibody tests. The ANA test is almost always positive when a human substrate is used (e.g., Hep-2 cells); in contrast, 5% to 10% of patients with SLE show a negative ANA test when nonhuman tissues are used for testing. Having a positive ANA test does not make a diagnosis of lupus. It can be seen in patients with several other collagen vascular disorders and is present in 5% to 10% of the normal population. However, an ANA level of greater than 1 : 160 usually suggests an autoimmune disorder. Five patterns of ANA have been described: speckled (the most nonspecific); homogeneous; peripheral or shaggy (most patients with SLE and this pattern have anti-ds [also called native ] DNA antibodies); nucleolar pattern (often seen in patients with scleroderma); and centromere pattern (largely associated with calcinosis, Raynaud, esophageal dysmotility, sclerodactyly, and telangiectasia [CREST]; see discussion on SSc).
Monitoring of serum complement is a critical test for determining lupus activity. The development of hypocomplementemia commonly signals the onset of renal disease. Total complement activity is monitored by functional hemolytic assay (CH50). Specific complement levels, particularly of C3 and C4, should be routinely checked by single radioimmunodiffusion assay. Not uncommonly, C4 levels may be depressed when C3 is normal, probably because patients with SLE are often missing one or more C4 alleles.
One risk factor for the development of SLE, especially with onset during childhood, is hereditary deficiency of the early complement components, C1 (C1q and less often C1r/s), C4, C2, and C3. Virtually all children with C1q deficiency develop SLE. The SLE that occurs in complete C1- or C4-deficient individuals typically presents early during childhood. C2-deficient individuals tend to have a lower risk and less severe disease with lower titers of antinuclear antibodies, but increased anti-Ro antibodies. Lupus and other autoimmune disorders do not tend to occur in individuals with deficiency of the later components of complement, which instead are associated with an increased risk of developing infections, including neisserial infections. Lupus has also been associated with patients and carriers of chronic granulomatous disease, and in patients with IgA deficiency.
Children who may have lupus should be tested for the presence of anti-ds DNA antibodies, which are found in about 50% of patients with SLE and have been associated with an increased risk of renal disease, particularly if complement levels are low. In contrast, antiribosomal P antibodies are inversely associated with renal involvement in juvenile-onset SLE. Anti-single-stranded (ss) DNA antibodies are found in about 70% of patients and are nonspecific. However, complement-fixing anti-ss DNA antibodies are associated with renal disease, even without anti-ds DNA antibodies. Some patients with SLE have antibodies directed against nuclear ribonuclear proteins (nRNPs). Patients with anti-nRNP antibodies (30% to 40%) have a lower risk of renal disease and a better prognosis. Anti-nRNP antibodies and a speckled ANA pattern have also been described in patients with an overlap syndrome of lupus in association with sclerodactyly, esophageal dysmotility, Raynaud phenomenon, and pulmonary disease (MCTD). The Raynaud phenomenon typically precedes the appearance of other signs and symptoms by several years in affected children. Fever, arthralgia, and myalgia are associated, and patients often have hypergammaglobulinemia, a positive RF, and normal complement levels. The anti-Smith (Sm) antibody is specific for SLE, occurs in 20% to 35% of patients, and is associated with a higher risk of renal disease. Anti-Ro and anti-La antibodies are less common in children than in adults (except in NLE, see Neonatal Lupus Erythematosus section) and occur in about 20% (anti-Ro) and 10% (anti-La) of children with SLE; they are associated with the highest risk of photosensitivity. SCLE is rare in children, but anti-Ro and anti-La antibodies can also be seen in children with SLE and complement deficiencies or Sjögren syndrome (see Sjögren Syndrome section). Antiphospholipid (aPL) antibodies can occur in up to 65% of children with SLE and should be monitored annually; they portend for the development of irreversible organ damage, not just a higher risk of thrombosis.
Other useful laboratory tests include the ESR as a measurement of inflammation; complete blood counts to detect leukopenia, thrombocytopenia, and a Coombs-positive hemolytic anemia; and routine urinalysis with blood urea nitrogen (BUN) and creatinine levels to detect renal abnormalities. Renal biopsy and serial blood pressure measurements are important tools for managing children with SLE and evidence of renal disease. If a patient with SLE has cytopenia and unexplained fever, the diagnosis of MAS, as described under JIA, must be considered. The disease is best recognized by hyperferritinemia and subsequently hypertriglyceridemia and hypofibrinogenemia.
Lupus may be triggered in pediatric patients by drugs, although the features tend to be milder and occurrence is considerably less than in adults, likely reflecting the more limited use of medications in children that are associated with the highest risk. These high-risk drugs are procainamide (occurrence 15% to 20%) and hydralazine (occurrence 5% to 8%). The cutaneous features of drug-induced lupus can be divided into those of SLE, SCLE, and CCLE or DLE) ( Table 22-2 ). More than 80 drugs can cause drug-induced lupus, especially with SLE-like manifestations. In drug-induced SLE, the typical cutaneous features of classic SLE are often absent (e.g., malar rash, discoid lesions, Raynaud phenomenon, mucosal ulcers, alopecia). Rather, photosensitivity, lesions involving the skin vasculature (livedo reticularis, palpable purpura, ulcers, bullae, urticaria and urticaria vasculitis) and erythema nodosum are most often described. The exception is reactions caused by TNF-α antagonists, which rarely cause drug-induced lupus but may manifest as the malar rash or discoid lesions.
| Skin Features | Noncutaneous Signs | Laboratory Findings | Most Common Pediatric Trigger(s) | |
|---|---|---|---|---|
| Drug-induced SLE | Photosensitivity, purpura, erythema nodosum, urticaria and urticarial vasculitis, necrotizing vasculitis Usually absent: malar or discoid rash (except TNF inhibitors), mucosal ulcers, alopecia, Raynaud phenomenon | Fever, arthralgias, myalgias, pericarditis, pleuritic, sometimes hepatitis Usually absent: CNS, renal, pulmonary involvement Hepatic changes from minocycline | ANA, antihistone antibodies, elevated ESR May be mild cytopenia | Minocycline, occasionally TNF-α inhibitors |
| Drug-induced SCLE | Annular polycyclic lesions and/or papulosquamous lesions, including on legs Less commonly, erythema multiforme-like or vesiculobullous lesions, necrotizing vasculitis | Usually no arthritis, serositis, or major organ involvement | ANA, anti-Ro, anti-La | Terbinafine, griseofulvin NSAIDs (piroxicam, naproxen) |
| Drug-induced CCLE | Discoid lesions in photosensitive distribution | Usually no other signs | ANA | TNF-α inhibitors |
The most common cause of drug-induced SLE in pediatric patients is minocycline, primarily used by adolescents with acne (see Chapter 8 ); overall the risk is low. This reactivity to minocycline is specific and has not been described in patients who are administered tetracycline or doxycycline. Patients usually experience malaise in association with myalgia, arthralgia, or arthritis. Livedo reticularis, antineutrophil antibodies, and elevation of hepatic transaminase levels have been described, but evidence of vasculitis, renal disease, and neurologic involvement are unexpected. Drug-induced lupus from minocycline occurs most commonly in female patients and at an average of 2 years after starting the medication. Affected individuals usually show a symmetric polyarthralgia or polyarthritis involving the small joints of the wrists and hands. Isoniazid, hydralazine, and procainamides may lead to the polyserositis of lupus but rarely cause cutaneous or renal disease. Drug-induced lupus is usually associated with increased titers of antihistone antibodies, a positive ANA level, and often anti-ss DNA antibodies. Anti-ds DNA antibodies are usually absent and complement levels tend to be normal.
Drug-induced SCLE shows the typical cutaneous features of SCLE, appearing at sun-exposed areas as annular polycyclic or papulosquamous lesions, but the legs are often involved, in contrast to classic SCLE. Disease usually develops 4 to 20 weeks after initiation of treatment. The most common drugs to trigger SCLE in children are antifungals (terbinafine and griseofulvin), but other groups are antihypertensives, calcium channel blockers, angiotensin converting enzyme (ACE) inhibitors, statins, and IFNs. Drug-induced CCLE is very rare, and usually results from use of TNF-α antagonists. Discontinuation of the offending agent is the most important intervention, with topical application of anti-inflammatory medications if needed and systemic corticosteroids if a vasculitis is present. Cutaneous changes may improve within weeks after drug discontinuation, but complete resolution may be prolonged over several months.
The prognosis for children with SLE has improved dramatically from nearly 100% mortality to a survival rate of greater than 90% because of earlier recognition and earlier and more aggressive therapy. The main causes of death in children are renal failure, CNS lupus, myocardial infarction, cardiac failure, or infection. Although life expectancy is increased, the sequelae of disease activity and administration of systemic medications cause considerable morbidity in 88% of patients. These include hypertension, growth retardation, chronic pulmonary impairment, premature atherosclerosis, ocular abnormalities, permanent renal damage, neuropsychiatric and neurocognitive impairment, osteoporosis, musculoskeletal damage, and gonadal impairment. Therapy for SLE currently depends on the extent of local and systemic involvement. Discoid lesions without SLE usually respond to topical anti-inflammatory medications and sun protection, but antimalarials and occasionally systemically administered corticosteroids may be needed for control. Pulsed dye laser has reduced inflammation for facial lesions refractory to topical anti-inflammatory medications and hydroxychloroquine (see next paragraph). Avoidance of exposure to sun and unshielded fluorescent light by wearing hats, sun-protective clothing, and appropriate broad-spectrum sunscreens with UVA as well as UVB protection is essential. These include sunscreens with a sun protection factor (SPF) of 30 or greater that contain good UVA blockers, such as titanium dioxide, zinc oxide, avobenzone, and/or the newer, more photostable UVA organic blockers (see Chapter 19 ). Cosmetic camouflage is an option for patients with residual dyspigmentation. Topical corticosteroids or calcineurin inhibitors (tacrolimus, pimecrolimus) are effective for most inflamed cutaneous lesions, although relatively potent formulations may be required.
Antimalarial therapy is considered first-line systemic therapy for cutaneous lupus and is beneficial for long-term suppression of the disease. In adults, hydroxychloroquine has been shown to delay disease onset, prevent flares, retard renal damage, and reduce autoantibody accumulation. For patients who are not helped by hydroxychloroquine monotherapy, quinacrine can be added without increased risk of retinal toxicity. In a study of adults with cutaneous lupus who were not successfully treated with hydroxychloroquine monotherapy, the addition of quinacrine 100 mg once daily to hydroxychloroquine produced significant clinical improvement in 67% of patients, as measured by reduction in the cutaneous lupus activity scale. According to the recommendations of the American Academy of Ophthalmology, patients treated with an antimalarial should have a pretreatment ophthalmologic examination and annual screening after 5 years, or sooner if there is an unusual risk factor, to detect early retinopathy. In addition to standard testing of the parafoveal area (10-2 field evaluation), at least one form of the newer, more sensitive objective testing (multifocal electroretinogram, spectral domain optical coherence tomography, or fundus autofluorescence) should be performed. The appropriate dosage of hydroxychloroquine for children is up to 6.5 mg/kg per day, with a maximum dosage of 400 mg/day. Several months of antimalarial therapy may be required before efficacy is seen.
For patients with severe refractory disease, thalidomide has proven efficacy in cutaneous lupus, although its side effect profile often limits routine use. Lenalidomide, a thalidomide analog, may also be used for severe scarring disease. A small study of patients with refractory CLE found that 86% achieved clinical remission with no influence on systemic disease. The drug is associated with fetal embryopathy, cytopenias, and an increased risk of thrombosis but, in contrast to thalidomide, not peripheral neuropathy.
Patients with mild disease and arthralgias without nephritis may respond to NSAIDs, although GI irritation is a risk. Although aspirin is generally not a treatment of choice for the arthralgias, 1 to 3 mg/kg per day or and 81 mg tablet is the recommended dosage for individuals with elevated levels of IgG aPL antibodies.
In patients with more than mild disease, corticosteroids are the mainstay of therapy. In general, children are treated more aggressively than adults, with 97% receiving systemic steroids and 66% receiving other immunosuppressive drugs. High dosages of systemic corticosteroids are usually given until complement and anti-ds DNA antibody levels normalize with improvement noted in the clinical state. High-dosage pulsed intravenous methylprednisolone (30 mg/kg) is generally administered for significant acute flares. Corticosteroids can then be administered orally and tapered slowly to the lowest possible level that controls the disease. Both alternate-day and single-morning dose therapy may be effective for maintenance, although alternate-day dosing is preferable to lower the risk of growth retardation. Avascular necrosis occurs more commonly in children than in adults (10% to 15%) from steroid usage. Children with SLE and immunosuppressive medications are at increased risk of infection, including pneumococcal infection; unfortunately the response to administration of pneumococcal vaccination has been poor. Patients should not receive live vaccinations during corticosteroid administration, and placement of a tuberculin test before starting therapy is recommended.
Given the association of chronic use of systemic corticosteroids with adverse effects on major organs (renal, cardiac, neurologic) and the recalcitrance of many patients to steroids alone, other immunosuppressive medications have also been administered to achieve or maintain disease control. The addition of an immunosuppressive agent to the corticosteroid has been shown to be more beneficial for patients with nephritis than the corticosteroid alone and allows a reduction of steroid dosage. Azathioprine, methotrexate, and, more recently, mycophenolate mofetil may reduce disease activity or allow lowering of the steroid dosage. The recommended pediatric dosage for azathioprine is 2.5 to 3.5 mg/kg per day. For methotrexate the dosage is 0.4 to 0.6 mg/kg per week with folate given daily or on days without methotrexate, and for mycophenolate mofetil, it is 0.5 to 3 g/day (600 to 1200 mg/m 2 per day). Cyclophosphamide use is most commonly reserved for patients with CNS disease or severe nephritis. The major potential side effects of these agents are hemorrhagic cystitis and sterility (cyclophosphamide); hepatitis (azathioprine); and aplastic anemia, infection, and malignancy (both cyclophosphamide and azathioprine). Mycophenolate mofetil has a better side-effect profile. Dapsone is of little value for SLE but can be quite effective for BSLE. Autoantibodies and polyclonal B-cell activation are thought to be important in the pathomechanism of SLE; targeting of specific B cells with rituximab (anti-CD20 chimeric mouse/human monoclonal antibody) has led to improvement, particularly for lupus nephritis and autoimmune cytopenia. Use of higher dosages than used for lymphoma (e.g., 750 mg/m 2 rather than 350 mg/m 2 ), repeated therapy, and combination therapy (e.g. with cyclophosphamide and corticosteroids) has led to improved disease activity and decreased steroid burden without an increased risk of infection requiring hospitalization. For end-stage renal disease, dialysis and transplantation are therapeutic options. Autologous stem-cell transplantation has been associated with significant morbidity and mortality, as well as high relapse rates in pediatric cases.
Levels of 1,25-dihydroxy vitamin D and intact parathyroid are significantly lower in children with SLE, especially if they are overweight. Preventive therapy for osteopenia in pediatric patients with SLE should include high dietary calcium intake with supplemental calcium and vitamin D (osteocalcin) as needed. Maintenance of a good exercise program and minimizing steroid weight gain are also helpful. Although bisphosphonates are not routinely given to pediatric patients, their use in patients with pathologic fractures secondary to steroid-induced osteoporosis should be considered.
Neonatal Lupus Erythematosus
Neonatal lupus eythematosus (NLE) is a unique variant of lupus erythematosus found in infants born to mothers with, or who have a tendency for, SLE, Sjögren syndrome, or undifferentiated autoimmune syndrome ( Box 22-2 ). The occurrence of NLE is not related to titers and is not strictly genetic, based on its discordance in identical twins. Nevertheless, developing the skin manifestations of NLE has been linked to carrying a polymorphism in the TNF-α receptor (308A; increases TNF-α expression in response to UV light) and the HLA genotypes DRB1*03. Cardiac block has been increased with DRB1*04 and Cw*05 and a polymorphism in TGF-β (Leu10, associated with increased fibrosis) ; DRB1*13, and Cw*06 are protective. The diagnosis of NLE is most important as a marker for the mother, who is at risk. Approximately half of mothers of affected infants are asymptomatic at the time of delivery, and about half of these originally asymptomatic mothers develop SLE, Sjögren syndrome, or, most often, undifferentiated autoimmune syndrome within a median of 3 years. In addition, mothers with undifferentiated disease may progress to reach a diagnosis of SLE, Sjögren syndrome, or overlap syndromes. Thus mothers of affected infants with NLE must be monitored and evaluated regularly.
- ▪
Seen in infants born to mothers with a tendency for systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or undifferentiated connective tissue disease
- ▪
May present as lupus-like rash, often in areas of sun exposure; facial erythema, especially periorbitally (raccoon eyes), annular lesions, discoid lesions, atrophic lesions, and/or telangiectasia
- ▪
Associated with anti-Ro (SS-A), anti-La (SS-B), and anti-U 1 RNP (nRNP) antibodies
- ▪
Congenital atrioventricular heart block in 15% to 30%
nRNP, Nuclear ribonuclear protein; RNP, ribonuclear protein.
Approximately 50% to 78% of babies with NLE show cutaneous manifestations at some point, with onset usually by a few weeks of age. However, up to 23% of affected babies show manifestations at birth, especially the cardiac manifestations and occasionally the cutaneous features. Typically, new lesions do not occur after approximately 3 months of age. Lesions are most commonly localized to sun-exposed areas, particularly on the head and neck, and the extensor surfaces of the arms, but they may occur anywhere on the skin surface. Lesions have been described in the genital area and on the palms and soles in 5% of patients.
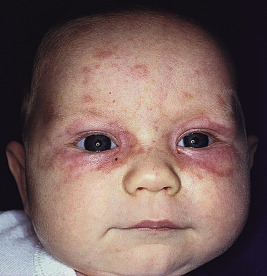
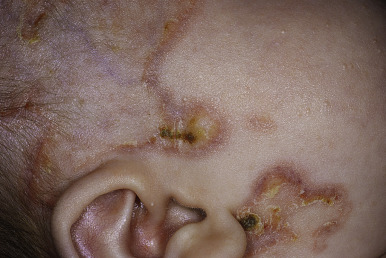
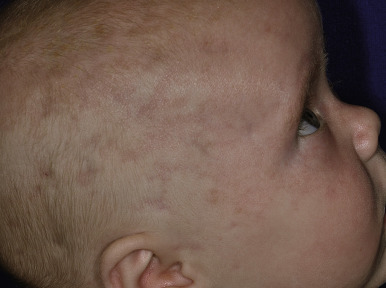
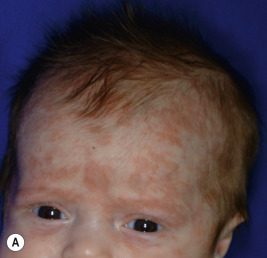
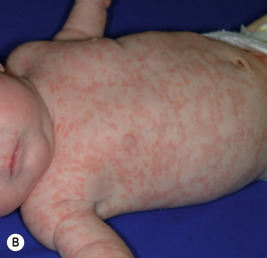
A variety of cutaneous manifestations have been described. Annular lesions or periorbital erythema (termed raccoon eyes, owl eyes, or eye mask ) ( Figs. 22-16 through 22-18 ) should always trigger consideration of the diagnosis in an infant. The annular lesions (see Fig. 22-17 ; Fig. 22-19 ) may be confused with annular erythema of infancy, Sweet syndrome, or neonatal dermatophyte infection. Discoid lesions ( Fig. 22-20 ), scaly atrophic macules and patches, and telangiectasia have also been described. Atrophic lesions may resemble round ice-pick scars, are often congenital, and commonly localize to the temporal areas of the forehead. The telangiectasia may be more petechial or may resemble the reticulated lesions of cutis marmorata telangiectatica congenita or an extensive capillary malformation. An infant with congenital varicelliform lesions has also been described. In addition to the cutaneous eruption, mucosal ulcerations have been noted in several affected babies.
NLE is the most common cause of congenital heart block. Congenital heart block occurs in 15% to 30% of affected patients, owing to in utero inflammation, fibrosis, and calcifications of the atrioventricular node and sometimes the sinoatrial node; 10% of patients with cardiac disease have cardiomyopathy. Although the cardiomyopathy is usually apparent early, rarely it first manifests several months after birth. Unlike the cutaneous complications of NLE, congenital heart block is usually irreversible. Some infants with first-degree and, rarely, with second-degree heart block have shown spontaneous resolution during the first few months of life, but the majority of patients progress rapidly to third-degree block, including after birth. Two-thirds of infants with heart block require pacemakers. Cardiac disease results in a 20% mortality not only from the heart block, but also congestive heart failure or other associated complications such as transposition of the great vessels, patent ductus arteriosus, septal defects, and endocardial fibroelastosis.
Other systemic complications include liver involvement in 10% to 26% of patients (generally manifested by hepatomegaly and abnormalities of liver function tests, but occasionally cholestatic disease and liver failure), splenomegaly, lymphadenopathy and hematologic abnormalities in 27% of affected infants, including leukopenia, thrombocytopenia, and anemia (both Coombs positive and Coombs negative). Subclinical evidence of CNS disease has been seen on computerized tomographic scans and ultrasounds, but clinical neurologic features are unusual. Hydrocephalus has recently been described as an uncommon complication, suggesting that imaging studies are appropriate for NLE infants with a head circumference greater than the 95th percentile. CNS vasculitis and stroke have also been described.
NLE results from the transplacental passage of maternal antibodies. In 95% of cases, these antibodies are anti-Ro (SS-A) antibodies, often in association with anti-La (SS-B) antibodies. These antibodies have been found at sites of pathology in the epidermis and heart. A subset of infants with neonatal lupus and their mothers have anti-U 1 RNP (nRNP) antibodies, although cardiac disease is extremely rare in this subset. Infants and mothers may also demonstrate a positive ANA titer and aPL antibodies. Breastfeeding has not been shown to alter antibody levels or affect the development of cutaneous lesions.
The anti-Ro antibodies have been shown to be causative for the cardiac block. Anti-Ro antibodies bind to fetal, but not to adult, cardiac myocytes and selectively injure the conducting system, a process that involves plasmin ; anti-U 1 RNP antibodies do not bind to cardiac myocytes. When pregnant mice are injected with anti-Ro/La antibodies intraperitoneally, the antibody binds to the heart, epidermis, and liver, sites of inflammation in NLE, and is associated with tissue apoptosis. Overall, 10% to 20% of babies of women with anti-Ro with/without anti-La antibodies develop cutaneous NLE; 1% to 2% develop heart block, and 27% show laboratory abnormalities.
The diagnosis of NLE is aided by the presence of typical cutaneous lesions, systemic manifestations, and confirmatory laboratory studies (positive anti-Ro, anti-La, or anti-U 1 RNP antibody tests of the infants and mothers). Because of the possibility of involvement of internal organs, a thorough physical examination, complete blood cell count with platelet count, and liver function tests are often recommended for infants suspected of having NLE. Infants with bradycardia or a murmur deserve an electrocardiogram and echocardiography. If any question regarding the diagnosis remains, cutaneous biopsy of a skin lesion can be performed and will show histopathologic features of lupus with injury to the basal epidermal cells as a prominent feature.
Clearance of cutaneous lesions generally occurs by 6 to 12 months of age, concurrent with the waning of the maternally derived antibodies. In a few patients, however, the cutaneous lesions disappeared within 1 month, and the eruption has lasted up to the age of 26 months. Affected individuals may show residual telangiectasia, dyspigmentation, atrophy, and/or scarring ; however, most cutaneous lesions clear without sequelae. Appropriate treatment of NLE includes avoidance of sun exposure and treatment of visceral complications, which may necessitate administration of systemic corticosteroid therapy. Low- to mid-potency topical steroids or topical calcineurin inhibitors can be used to treat the cutaneous lesions, but lesions clear spontaneously, and the use of topical anti-inflammatory agents has not been shown to affect residua.
Patients with NLE do not show an increased risk of developing lupus and other autoimmune disorders beyond that of their siblings, suggesting that the small increased risk in this group of patients reflects their familial tendency, not the previous occurrence of NLE. Mothers who have had a child with NLE, however, have a 36% overall risk of having a second affected child. The risk of cutaneous manifestations in the second child is 23%, and the risk of cardiac issues is almost 13%, sixfold higher than the overall risk congenital heart block in a first affected baby. A mother with a previous baby with NLE involving skin can have a subsequent baby with congenital heart block and vice versa. Fetal echocardiograms (weekly to every other week) between 16 and 26 weeks’ gestational age, the peak period of cardiac injury, are recommended for at-risk pregnancies. Systemic corticosteroids administered during pregnancy may decrease the risk of first- or second-degree heart block but have not been shown to prevent the progression toward third-degree heart block, which can occur quickly in utero . Antimalarials, particularly hydroxychloroquine, are not teratogenic and have been found to decrease the risk of cardiac, although not noncardiac, manifestations of NLE. The combination of steroids with intravenous Ig and plasmapheresis is under investigation.
Antiphospholipid Antibody Syndrome
Antiphospholipid antibody syndrome (APS) is an autoimmune thrombotic disorder, characterized by venous and/or arterial thrombosis and the persistence of at least one circulating aPL antibody. aPLs are a heterogeneous group of autoantibodies reactive against either negatively charged phospholipids (e.g. phosphatidyl serine and cardiolipin) or proteins that are complexed with them (e.g., β2 glycoprotein I). aPL antibodies are commonly found after bacterial and viral infections in children, leading to the requirement that two levels of aPL antibodies be tested at least 12 weeks apart for diagnosis.
In approximately half of affected pediatric patients, APS is a primary syndrome. The disorder can also occur secondarily, most commonly in children with SLE (38% of children overall). However, 30% of the patients with SLE and APS initially have primary APS, with a mean duration of 1.2 years before the onset of SLE. The age of onset is significantly younger in children with primary APS (8.7 vs. 12.7 years), and 45% of tested children also have one or more inherited thrombophilic risk factors. These inherited prothrombotic disorders include methylenetetrahydrofolate reductase C677T polymorphism, factor V Leiden, protein S or protein C deficiency, prothrombin G20210A heterozygosity, and antithrombin III deficiency.
Manifestations may range from headache or livedo reticularis to stroke or severe tissue necrosis ( Box 22-3 ). The most common cutaneous manifestations of APS in children are livedo reticularis and Raynaud phenomenon, each occurring in approximately 6% of affected children. Livedo reticularis is characterized by prominent reticulated cutaneous vasculature, most commonly noted on the lower extremities (see Fig. 22-10 and Chapter 12 , Fig. 12-81 ). Thrombosis, is the most serious manifestation (see Box 22-3 ), and venous thromboses involving the lower extremities occur most commonly. Recurrent thromboses occur in approximately 20% of children, especially with inadequate anticoagulant therapy and with an underlying inherited thrombophilia. The clinical triad of livedo reticularis, hypertension, and cerebrovascular disease, or Sneddon syndrome, is linked to aPL antibodies in individuals and has rarely been described in children.
Cutaneous and Subcutaneous Features
Livedo reticularis
Raynaud phenomenon
Palpable purpura/leukocytoclastic vasculitis
Superficial vein thrombosis
Deep venous thrombosis with ulceration
Pyoderma gangrenosum-like ulceration
Degos-like disease
Gangrene
Noncutaneous Features *
* Venous thromboses occur in 60% of patients; arterial thromboses in 32% (45% of patients with primary antiphospholipid antibody syndrome [APS] and 18% with secondary APS); mixed thromboses in 3% of patients; and small vessel thromboses, particularly leading to digital infarctions in 6% of patients.
Thromboses *
Pulmonary thromboembolism, hypertension
Strokes, transient ischemic attacks
Transverse myelopathy
Cerebral venous sinus thrombosis
Retinal vein thrombosis
Renal artery thrombosis
Hematologic abnormalities (38%)
Evans syndrome
Thrombocytopenia
Leukocytopenia
Autoimmune hemolytic anemia
Neurologic abnormalities, nonthrombotic (16%)
Migraine headaches
Chorea
Seizures
Hepatomegaly, enzyme elevation
Five percent of children have life-threatening thrombotic disease with the rapid occurrence of thrombosis involving at least three organ systems (catastrophic APS). Affected children are likely to be female and suffer from primary APS (each about 70%). The catastrophic event is often the first sign of APS in children (87%). Infections are often a precipitating factor (61%) and peripheral vessel thrombosis occurs in 52% of affected children.
aPL antibodies have been associated with a high risk of miscarriage in pregnant women. In successful pregnancy, transplacental passage of maternal aPL antibodies can rarely lead to neonatal thrombosis, most commonly manifesting as stroke. The risk may be increased in aPL-positive neonates by acquired thrombic risk factors such as central lines, sepsis, prematurity and heart disease, or the hereditary prothrombic diseases previously mentioned. Neonates with stroke may have persistently elevated aPL titers for years that show no concordance with maternal aPL levels.
aPL antibodies in APS are thought to be pathogenic, rather than merely a serologic marker. Anticardiolipin, lupus anticoagulant, and anti-β2 glycoprotein I antibodies are found in 81%, 72%, and 67% of patients, respectively, without a significant difference between primary and secondary types of APS. Only 33% of pediatric patients have all three aPL subtypes, and 48% have two simultaneously, emphasizing the need to evaluate all three antibodies for diagnosis. Of note, having more than one aPL antibody does not increase the risk of thrombosis. ANAs and anti-ds DNA antibodies are detected much more commonly in children with secondary APS (86% vs. 35% and 59% vs. 6%, respectively).
Despite the established association between aPL antibodies and thromboses, most individuals with circulating aPL antibodies do not show evidence of thrombosis. As such, it remains controversial whether to treat individuals prophylactically who show persistently positive aPL antibody levels but have no history of thromboses. There is currently no evidence to support the routine administration of low-dose aspirin in individuals with IgM or low titers of IgG antibodies; however, anticoagulants (e.g., aspirin at 1 to 3 mg/kg per day or an 81 mg tablet) should be considered in patients with higher levels of aPL antibodies, especially patients who will be immobilized or undergoing surgery. Administration of hydroxychloroquine may be protective against the development of thrombosis in aPL+ children with SLE. In addition, at-risk adolescents should be discouraged from smoking or treatment with estrogen-containing oral contraceptives. Children with aPL antibodies and thrombosis require anticoagulant therapy, but the use of these medications should be weighed in very active children and noncompliant adolescents because of the greater risk of hemorrhage. Children with catastrophic APS are treated with intravenous anticoagulation, plasmapheresis, and sometimes rituximab.
Degos Disease
Degos disease, also known as malignant atrophic papulosis, is an often-fatal disorder characterized by multiple infarcts in the skin and viscera owing to a thrombotic vasculopathy of unknown cause. Altered platelet function or fibrinolysis have been noted in some patients. The condition is particularly rare in children but has been reported to occur in infants as young as 1 day of age. Typical lesions are asymptomatic 3- to 10-mm papules with an atrophic, porcelain-white center surrounded by an erythematous, telangiectatic border ( Fig. 22-21 ). Lesions may appear in crops and typically involve the trunk and extremities; one adolescent with cutaneous Degos-like lesions has been described with localized unilateral involvement of the abdomen. Biopsy of lesional skin shows a characteristic acellular, ischemic wedge-shaped area of dermis underlying an atrophic but hyperkeratotic epidermis. The GI tract and CNS are the most commonly affected organs. Although involvement of the GI tract is most typical, of the 25 reported pediatric cases of Degos disease, 56% showed CNS involvement and 48% had GI involvement. Acute hematemesis, abdominal pain, dyspepsia, abdominal distension, diarrhea, and constipation are signs of GI tract involvement, and patients may develop peritonitis, bowel infection, or perforation. CNS manifestations include fatal hemorrhagic or ischemic strokes, polyradiculoneuropathy, and nonspecific neurologic symptoms, especially headache. In most cases, the skin lesions precede the occurrence of other manifestations by months to years. Most affected patients die within 2 to 3 years, particularly from sepsis from peritonitis, CNS bleeding, or pleural or pericardial involvement. Approximately 36% of affected children have a more benign course without evidence of systemic involvement, in contrast to 15% of patients of all ages. Familial cases have been described. Degos-like lesions have been described in patients with aPL antibodies and/or SLE and, less commonly, dermatomyositis, suggesting that Degos disease is a distinctive disease pattern rather than a unique entity. In patients with SLE and dermatomyositis, the lesions tend to be more telangiectatic and show more mucin deposition. Administration of medications that inhibit increased platelet aggregation (especially aspirin and dipyridamole) may be helpful in some cases. Immunosuppression may worsen the skin lesions of Degos disease.
Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is an inflammatory disorder with an incidence of three per million children per year that primarily affects the skin, striated muscles, and occasionally other internal organs ( Box 22-4 ). In cases in which cutaneous changes are absent, the term polymyositis is used. Because the clinical and pathologic features of involved skin and muscles are similar, dermatomyositis and polymyositis are believed to be variants of the same disease process; however, 85% to 95% of children with an inflammatory myopathy have JDM.
General
Fever
Lethargy
Adenopathy
Skin, Subcutaneous, Mucosae
Heliotrope rash
Gottron papules
Nailfold capillary changes
Malar or facial eruption
Mouth ulcers
Gingival telangiectasia, bleeding gingivae
Skin ulcers
Limb edema
Xerosis and pruritus of skin, including scalp
Poikiloderma
Calcinosis
Lipodystrophy
Muscle
Muscle tenderness and myalgias
Muscle weakness
Joints
Arthralgia
Arthritis
Contractures
Gastrointestinal
Dysphagia/dysphonia
Gastrointestinal symptoms, especially pain
Pulmonary
Dyspnea from interstitial lung disease
Cardiac
Murmurs, cardiomegaly, pericarditis
Rarely, myocarditis, conduction abnormalities
The criteria to make a diagnosis of JDM were originally described in 1975 and remain largely the same today, except that electromyographs, which are painful and technically challenging, have largely been replaced by magnetic resonance imaging (MRI) evaluation ( Box 22-5 ). Muscle biopsies, although performed to diagnose the disease in only 61% of patients, provide important diagnostic and prognostic information and are critical if the cutaneous features are not typical.
- ▪
Progressive symmetric weakness of the proximal muscles (limb girdle and anterior neck flexors), with or without dysphagia or respiratory weakness
- ▪
Muscle biopsy evidence of myositis and necrosis
- ▪
Elevation of muscle-derived serum enzyme levels
- ▪
Evidence of myopathy on electromyography/magnetic resonance imaging
- ▪
Typical cutaneous findings of dermatomyositis
A definitive diagnosis of dermatomyositis can be made when specific skin findings and three of the above criteria (excluding the rash) are present; a probable diagnosis of dermatomyositis requires two of the above criteria plus the rash.
JDM occurs more often in girls than boys (2.3 : 1) and shows a bimodal age distribution, with one peak between 2 and 5 years of age and another at 12 to 13 years of age, giving a mean age of onset of 7 years of age; 35% of children have the onset of JDM before 5 years of age. Overall approximately 25% of all individuals afflicted with dermatomyositis are younger than 18 years of age at the time of onset. Malignancy is associated with adult dermatomyositis in about 20% of cases but is rare in pediatric patients. In contrast to adults, children tend to have more inflammation and necrosis of muscle, calcinosis, periungual and gingival telangiectasias, ulceration, and small vessel vasculopathy, but fewer myositis-specific antibodies, less interstitial lung disease and amyopathic disease, and a lower mortality rate. The histologic features in muscle and IFN gene signature of JDM are similar in both adult and childhood disease.
The immune alterations that drive the small-vessel occlusive vasculopathy of JDM are still poorly understood, but recent studies suggest important roles for environmental triggers, immune dysfunction, and specific tissue responses. Although the onset shows seasonality and many children have preceding infectious features, no evidence of an associated infectious agent has been discovered. Maternal microchimerism has also been implicated in causing JDM, and has been noted in 70% of peripheral blood T lymphocytes and 80% to 100% of muscle tissue samples from patients. The major histocompatibility complex alleles HLAB8, HLADQA1*0501, HLADQA1*0301, HLADRB*0301, and HLADPB1*0101 confer increased risk. In addition, several other polymorphic gene loci have been shown to be risk factors. A polymorphic variant of the TNF-α promoter that increases TNF-α expression (TNF-308A), including in response to UV light, is associated with a higher risk of calcinosis, ulcerations, and disease chronicity. Polymorphisms in IL-1 and the IL-1 receptor antagonist also increase the risk of the development of calcinosis and JDM, respectively.
The onset of JDM is insidious in approximately 50% of affected children with subtle weakness, anorexia, malaise, abdominal pain, and the gradual development of a rash. In about 30% of children, the onset of the disorder is fulminant, with fever, profound weakness, and severe multisystemic involvement. The remainder have a subacute onset, often with the cutaneous eruption preceding the appearance of constitutional symptoms or evidence of myopathy by 3 to 6 months, although occasionally by as long as several years; hypomyopathic disease occurs in the presence of skin signs but subclinical muscle disease. An amyopathic form, characterized by typical cutaneous manifestations without clinical or laboratory evidence of myositis for at least 6 months, is less common in children than in adults. In one study, 26% of these children with “amyopathic JDM” develop evidence of myositis during a mean follow-up period of 3.9 years, and the 4% who developed calcinosis all developed myositis, suggesting that children without myositis can be treated with antimalarials and topical anti-inflammatory medications, but may not need aggressive immunosuppressive management. However, given the known increased risk of calcinosis without early intervention, there is continued controversy about management of amyopathic disease.
The cutaneous features of JDM may be striking or so minor that they might be easily overlooked. A variety of cutaneous findings maybe noted (see Box 22-4 ). Characteristic cutaneous lesions are inflammatory and telangiectatic and are found in 75% of affected children at presentation. A purplish-red erythema occurs on the face, especially on the eyelids, and is called the heliotrope rash, because it matches the color of reddish purple flower of the heliotrope plant ( Fig. 22-22 ). The eruption can also be seen on the upper cheeks, forehead, temples, and ears ( Fig. 22-23 ). Sometimes the facial lesions are very edematous, leading to a periorbital and cheek edema. The facial edema and discoloration may be asymmetric, reflecting asymmetric disease activity; JDM has also first manifested in a Blaschkoid facial distribution with later involvement in a generalized distribution. Violaceous, often confluent, telangiectatic erythema with fine scaling may also appear at the hairline of the scalp, nape of the neck, extensor surfaces of the arms and shoulders, the elbows and knees, and the shoulder and center of the upper anterior trunk (the shawl sign) ( Fig. 22-24 ). These telangiectatic eruptions are often accentuated when the affected area is dependent (e.g., with lowering the face for facial lesions). The distribution of the eruption and the common sparing of sun-protected areas suggest photosensitivity; UV light exposure is known to trigger both cutaneous and muscle signs.
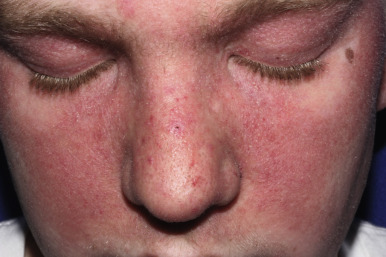
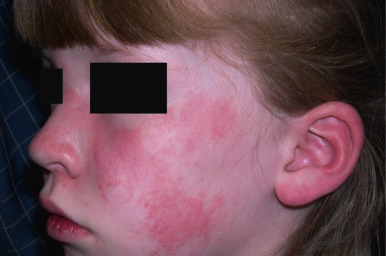
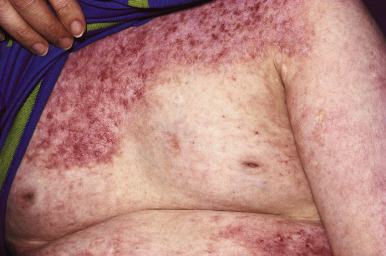
Periungual telangiectasia ( Fig. 22-25 ) is a characteristic feature of JDM and provides a good indication of cutaneous, rather than muscle, disease activity. Viewing the nailfold area through a dermatoscope, ophthalmoscope, or otoscope to magnify the area after local application of mineral oil improves visualization. Nailfold capillary microscopy has shown that the periungual telangiectasia represents bushy capillary loops adjacent to avascular areas, perhaps a form of compensatory neovascularization. Erythema of the cuticle at the base of the nails commonly accompanies the periungual telangiectasia. At times, the cuticles may also be thickened, hyperkeratotic, and irregular, giving an appearance of excessive picking or manicuring. Pitted ulcerations of the fingertips, pressure points such as the elbows, lateral aspects of the trunk, axillae, and lateral ocular canthi are usually seen at presentation or with disease flares. Thought to be a marker of severe disease, these ulcers leave residual atrophic scars. Even though not exposed to sun, the mucous membranes often show telangiectatic involvement, with erythema of the palate and buccal mucosa, with or without ulceration, and telangiectasia of the gum margin ( Fig. 22-26 ).
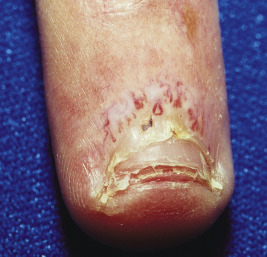
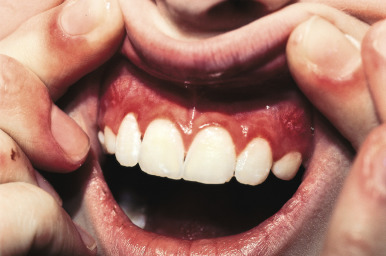
Telangiectatic erythema may overlie the dorsal interphalangeal joints (Gottron sign), and when a component of a flat-topped papule, is a pathognomonic sign of JDM (Gottron papule) ( Fig. 22-27 ). Coupled with the facial eruption, these inflammatory lesions on the dorsum of the hand may be misdiagnosed as contact dermatitis. When the papule resolves, atrophy, telangiectasia, and hypopigmentation may persist ( Fig. 22-28 ). Inverse Gottron papules have also been described with papules on the palmar surface; in one report these were the only cutaneous feature and were associated with interstitial lung disease.
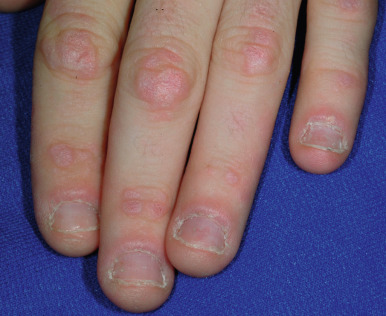
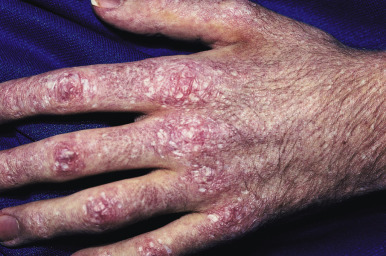
Other cutaneous features include palmar erythema and thickening, which can be mistaken for dermatitis and sometimes has so much hyperkeratosis and fissuring that it has been compared with a mechanic’s hand. A more generalized hyperkeratotic disorder that resembles pityriasis rubra pilaris, and generalized erythroderma with scaling, have also been described.
Sclerodactyly may be seen in children with overlap syndrome that includes SSc. Panniculitis may manifest clinically as tender, indurated plaques and nodules, especially on the arms, thighs, and buttocks; however, panniculitis is often observed in biopsy specimens and is not noted clinically. Limb edema is noted in some patients at presentation, but anasarca is rare and should prompt consideration of nephritic syndrome.
Several cutaneous features of JDM are seen more commonly with disease chronicity. Many children and adolescents with chronic disease have intensely pruritic, xerotic skin that shows poikiloderma ( Fig. 22-29 ), which is the combination of cutaneous atrophy, telangiectasia, and dyspigmentation (see Chapter 11 ). The scalp is dry, scaling, and pruritic as well.
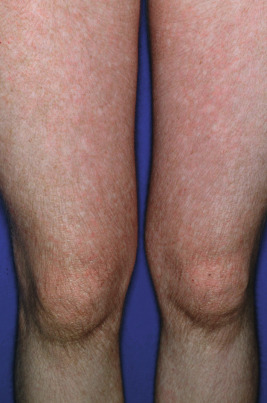
Cutaneous calcinosis develops in 18% to 24% of children, in contrast to the up to 70% incidence of earlier reports. This decreased incidence of calcinosis can be attributed to both earlier diagnosis and to more aggressive, earlier treatment (see below). Nevertheless, calcinosis is a major cause of morbidity in JDM and is linked to younger age at disease onset, more persistent disease activity, and the presence of anti-NXP2 (anti-p140) autoantibodies before the calcinosis develops, which are found overall in 20% of JDM patients. Most commonly seen at sites of trauma, particularly on the buttocks, elbows, knees, and fingers, and around the shoulders, subcutaneous calcium deposits may produce local pain and can be extruded, leading to ulcers, sinuses, or cellulitis ( Fig. 22-30 ). Ulcerations may also appear in children with dermatomyositis without calcifications, especially over joints ( Fig. 22-31 ). Calcifications (see Chapters 9 and 23 ) appear as hard, irregular nodules that may drain a whitish, chalky material through their openings to the surface that resembles gritty toothpaste. The mean time to occurrence of calcinosis after onset is 2.5 to 3.4 years, although 3% of patients have calcinosis at diagnosis, even without evidence of myositis. Sometimes patients have widespread calcification along fascial planes, encasing muscles (universalis form) or even appearing as an exoskeleton on radiographic examination.
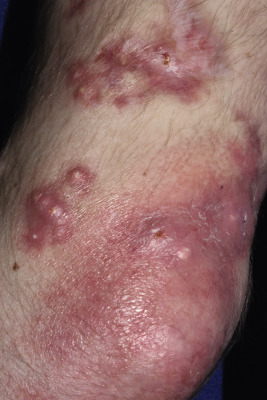
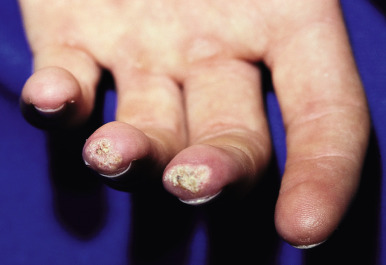
Contractures have been associated with the calcinosis and occur in 17% to 30% of patients with JDM. Lipodystrophy is a common feature in children with chronic disease, especially if poorly controlled and of longer duration, overall occurring in 9.7% of patients ( Figs. 22-32 and 22-33 ). The lipodystrophy may be generalized or partial and is characterized by a slowly progressive symmetric loss. The lipodystrophy is associated with insulin resistance and hyperlipidemia (hypertriglyceridemia and low levels of high-density lipoprotein [HDL]). These cardiac risk factors can be present in patients with chronic disease, even without the lipodystrophy. Acanthosis nigricans has been described in 10% of patients later in the disease course ( Fig. 22-34 ). The insulin resistance correlates with muscle atrophy, proinflammatory circulating cytokines, and a family history of diabetes, but not with corticosteroid use.
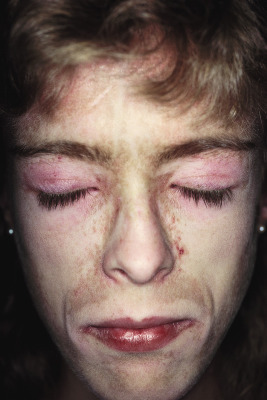
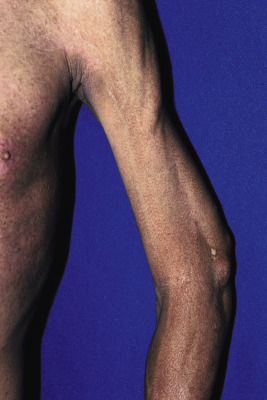
The myopathy of JDM affects primarily the proximal groups of muscles, especially the triceps and quadriceps, usually in a symmetric distribution. Muscle tenderness or stiffness is not uncommon but usually occurs later in the course rather than as a presenting sign. Affected children often complain of great fatigue and are unable to do simple tasks, such as rising, squatting, walking, or reaching. In young children, muscle weakness may manifest as more demanding behavior. Physical therapists may be helpful as assessors of muscle strength, particularly in younger children. Speech may become nasal because of nasopharyngeal muscle weakness, and approximately 10% to 15% of patients complain of difficulty with swallowing.
A nondestructive, generally nondeforming arthritis occurs in approximately half of affected children at some time during the disorder, most commonly an asymptomatic arthritis of the knees; large and small joint polyarthritis has also been described. Vasculopathy of the GI tract may lead to ulceration, perforation, pneumatosis intestinalis, or hemorrhage. D-xylose absorption has been decreased in patients with JDM, suggesting the decreased absorption of nutrients and medications, including oral corticosteroids, and leading to the suggestion that the disorder be treated with intravenous methylprednisolone for acute cases. Dysphagia from impaired muscle function may lead to esophageal reflux and aspiration pneumonia, requiring intravenous feeding. Children with JDM have occasionally been described with hepatomegaly, cholestasis, or pancreatitis.
The majority of patients with JDM show a reduced ventilatory capacity without respiratory complaints, probably owing to respiratory muscle weakness and poor chest wall compliance. Decreased diffusion capacity and evidence of interstitial lung disease is uncommon, but may present with dyspnea on exertion. Cardiac disease is rare in children with JDM, and mainly involves murmurs and cardiomegaly. Although pericarditis occurs, conduction defects and acute myocarditis are uncommon but severely damaging. Nevertheless, systolic dysfunction has recently been noted after long-term follow-up study (16.8 years) and is correlated with skin disease scores, not muscle disease activity. Central or peripheral nervous system disease is very rare, in contrast with SLE. Similarly, other than conjunctival vessel tortuosity, eye disease is usually limited to cataracts from administration of systemic corticosteroids. IgA nephropathy has been described, but renal disease is not generally a feature of JDM.
The presence of arthritis, significant dysphagia, renal disease, Raynaud phenomenon, or neuropathy should also prompt consideration of evaluation for overlap disease. Up to 10% of children with JDM show an overlap syndrome with features of SLE, SSc, or JIA.
The diagnosis of JDM is suspected on clinical grounds, based on the typical cutaneous manifestations and evidence of muscle weakness and/or tenderness. Increased levels of muscle enzymes and the demonstration of increased signal intensity on fat-suppressed T2-weighted MRI scans provide evidence of muscle inflammation, and whole body MRIs have been advocated to estimate total inflammatory burden. The increased signal intensity on MRI scans relates to accumulated extracellular water content. It is indistinguishable from other inflammatory myopathies or rhabdomyolysis but can be differentiated from muscular dystrophies. Several enzyme levels should be assessed concomitantly (serum aldolase, aspartate aminotransferase, lactic dehydrogenase, and serum creatine phosphokinase levels); aldolase is most commonly elevated and serum creatine phosphokinase is normal in about 35% of affected children with active disease. Muscle biopsy of a site shown to be affected by clinical examination or MRI testing can be performed to confirm the presence of perifascicular and perivascular lymphocytic infiltration with atrophy, necrosis, and late fibrosis of the muscle fibers. Skin biopsy is usually not valuable.
Children with active JDM are often lymphopenic but show a relative increase in the percentage of B cells, as defined by anti-CD19 monoclonal antibody binding. Overall, 70% of children with JDM have one or more detectable myositis-specific and myositis-associated antibody if full serologic testing is performed. The high incidence in adults of myositis-specific antibodies, particularly anti-Jo-1, is a distinguishing feature between adult and pediatric disease. Indeed, anti-Jo-1 antibodies (against histidyl-tRNA synthetase) are found in fewer than 3% of patients with JDM, and anti-Mi-2 antibodies (against a DNA helicase) have been found in about 5%. However, other more recently recognized autoantibodies targeting intracellular proteins (anti-TIF1-γ, anti-NXP2, and anti-MDA5) have been found in JDM and are useful biomarkers for defining distinct clinical subsets and predicting prognosis. Anti- TIF1-γ (p155) antibodies are detected in approximately 25% of children and are linked to more severe cutaneous involvement with generalized lipodystrophy. Anti-NXP2 (also called anti-MJ or anti-p140 ) antibodies, found in about 20% of patients, are associated with a substantially increased risk of greater disease severity, and of developing calcinosis and contractures. Anti-MDA5 (also called anti-CADM ) antibodies, noted in 7.4% of JDM patients, predict a distinct phenotype that includes skin and oral ulcers, arthritis, milder and shorter duration muscle disease activity, and, in a minority of patients, interstitial lung disease.
Children with overlap disease may have anti-PM-Scl (5% to 7%) or anti-U 1 RNP (<6%) antibodies. ANAs are also present in 60% to 70% of newly diagnosed children, usually in a coarse speckled pattern.
The course of JDM can be monophasic (lasting remission within 1 or 2 years, 37%), chronic (60%), or polyphasic (3%). Of JDM patients observed into adulthood (median time from diagnosis, 16.8 years), 90% of patients had persistent damage to the skin (77%), muscle (65%), and skeletal (57%) systems. Unfortunately, prediction of disease course has been a challenge, although inadequate treatment is a strong predictor of poor outcome. A shorter duration of untreated disease and a lower JDM skin disease activity score at onset of disease are correlated with a monocyclic course. The presence of persistent skin eruptions (especially Gottron papules) 3 months after initiation of treatment predicts a longer time to remission, and if Gottron papules and nailfold capillaroscopy are both abnormal at 6 months after initiation, the outcome is particularly poor. Organ damage 6 months after onset of treatment is also a predictor for poor prognosis. By 36 weeks, persistent nailfold capillary changes, which correlate with skin disease rather than muscle disease, also predict a nonmonocyclic disease.
During the active phase of dermatomyositis, children are particularly at risk for sudden, overwhelming Gram-negative sepsis, sometimes masked by corticosteroids. Hospitalization may be advisable in the acute stage, especially if the disease involves palatal-respiratory muscles, to prevent aspiration and to ensure adequate respiration, including by feeding intravenously and using mechanical ventilation if necessary. A high index of suspicion must be maintained for aspiration because of weak palatal-respiratory function or perforation of a viscus, and early aggressive therapy must be instituted when necessary. Nevertheless, the mortality rate, formerly about 30%, generally because of these complications, has been markedly reduced by intervention, and most recently was found to be 3% to 4%.
The course of JDM can be classified as monophasic in 47%, polyphasic in 18%, and chronic in 35%. The Pediatric Rheumatology International Trials Organisation (PRINTO) criteria and a Myositis Disease Activity Assessment Tool (MDAAT) have been developed to define disease activity versus inactivity. In one cohort with a median follow-up period of 16.8 years after symptom onset, 50% to 73% of patients had continuing disease activity, most commonly in the skin (59%).
Most JDM is treated with high-dosage corticosteroid therapy, and many specialists now initiate pulsed intravenous steroids (30 mg/kg or up to 1.5 g), administered every 24 to 48 hours until laboratory values normalize. Usage of pulse steroids, followed by oral corticosteroids or intermittent pulses if needed for continued control, has been shown to decrease the duration of rash, cause less functional impairment, and markedly reduce calcifications in comparison with those children treated with oral corticosteroids only. Side effects are not increased with pulse steroids, and the overall dosage of required corticosteroids tends to be less than that with oral treatment.
Some patients may fail to respond even to very large doses of corticosteroids. Methotrexate is generally the second-line treatment for JDM, when the disease is recalcitrant or corticosteroid usage is problematic. Treatment of severe JDM early in the course with both intravenous methylprednisolone and methotrexate appears to be more useful than with either agent alone, and patients with milder cases have responded to methotrexate (or intravenous immunoglobulin [IVIG]) alone. Methotrexate can be administered orally or, preferably, by subcutaneous injection at a dosage of 1 mg/kg per week with a maximum of 40 mg/week. Intravenous gammaglobulin has also been found to be an effective steroid-sparing agent, especially for cutaneous manifestations. Other cytotoxic agents such as azathioprine in dosages of 1 to 3 mg/kg (up to 200 mg/day) or cyclosporine (5 mg/kg per day) may be used in severe life-threatening forms of dermatomyositis, or when the disease cannot be adequately managed with prednisone alone. However, biologics, and particularly rituximab, are showing promise. Rituximab has been generally well tolerated, with infections and infusion reactions as the main adverse events. Although the onset of action can be slow, the response is good, including with more than one course. TNF-α has been implicated in the pathogenesis and severity of JDM, and some patients have shown benefit, especially with administration of infliximab ; however, other patients have not responded, particularly with etanercept, or have even worsened on a TNF-α antagonist. Disease that is refractory to immunosuppressants, including rituximab, can be treated with autologous stem-cell transplantation.
The response of the cutaneous features of JDM to treatment may not correlate with the response of muscle; skin manifestations may persist for years despite resolution of other signs of disease. Oral antimalarial agents, especially hydroxychloroquine (generally 5 to 6.5 mg/kg per day), have been helpful in controlling the cutaneous manifestations of dermatomyositis. Topical corticosteroids and calcineurin inhibitors do little to reverse the skin signs of JDM, although topical fluocinolone 0.01% in oil has been helpful with the pruritus of chronic scalp involvement. Counseling about sun protection is critical (see discussion for SLE).
The problematic calcinoses may gradually disappear but can cause considerable morbidity. Despite the long list of medications that have been used successfully in anecdotal reports in an effort to hasten clearance, no agent has been helpful in the majority of cases. Nevertheless, bisphosphonates in combination with aggressive immunosuppression have led to dramatic improvement in some patients. Sodium thiosulfate can help clear calcinoses when given intravenously, orally, or even topically, but it is often poorly tolerated (e.g., oral administration may lead to severe diarrhea). The combination of abatacept and sodium thiosulfate reduced the muscle and ulcerative skin disease activity, as well as calcifications.
Surgical procedures may also be useful in selected situations. Occlusive dressings (such as DuoDerm) over joint areas, particularly when areas are ulcerated and draining, have promoted healing. Physiotherapy is important for patients with or at risk for contractures to prevent deformity and increase muscle strength. MRI evaluations, myometry, and blood studies did not show increased muscle inflammation after exercise, suggesting that a moderate exercise program is acceptable with active myopathy.
Related posts:
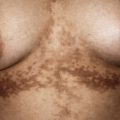
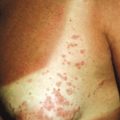
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


