Clinical criteria
Vascular thrombosis
Venous or arterial thrombosis may be present
Thrombosis must be confirmed by imaging studies or by histopathology
Laboratory criteriaa
1. Anti-cardiolipin antibody
2. Anti-β2 glycoprotein-1 antibody
3. Lupus anticoagulant
Investigation Recommended
Laboratory investigations | Antiphospholipid panel, including anti-cardiolipin antibody, anti-β2 glycoprotein-1 antibody, and lupus anticoagulant |
ANA and ENA panel to rule out underlying autoimmune condition | |
Hypercoagulability panel to assess for inherited thromobophilia risk factors | |
Imaging studies | Venous duplex if venous thrombosis suspected |
CT or MRI if arterial thrombosis suspected | |
Skin biopsy | If small vessel thrombosis suspected |
Anticoagulation is the mainstay of treatment for a thrombotic event in APS, and does not differ substantially from treatment of a patient without APS. Unfractionated heparin or low molecular weight heparin is typically used as a bridge to warfarin therapy, with a goal INR of 2.0–3.0. In the case of an arterial thrombosis, low-dose aspirin therapy, with or without concomitant warfarin, is recommended [2, 5]. Approximately 20 % of pediatric patients with APS have been documented to have recurrent thrombosis after their initial presentation [1, 5], so long-term anticoagulation and/or antiplatelet therapy is likely to be useful for prevention of recurrent events. The recommended duration of treatment has not been established, however, and the benefits must be weighed against the risk of hemorrhage, especially in active children [2]. In life-threatening situations, such as catastrophic APS, more aggressive treatments such as high-dose systemic steroids, plasmapheresis, intravenous immunoglobulin (IVIg), cyclophosphamide, or rituximab can be considered [2–4, 9]. Of note, precipitating factors such as infection, immobilization, and surgery are commonly seen in patients who develop thrombotic events, and these risk factors should be treated proactively [5, 10].
Table 22.2
First line therapies
Therapy | Dose | Evidence level |
|---|---|---|
Heparin bridge to warfarin (for acute thrombotic event) | Titrate to goal INR 2–3 | C |
Aspirin (for arterial thrombotic event) | 3–5 mg/kg per day, max 325 mg per day | D |
Long-term warfarin ± aspirin | Goal INR 2–3, duration and intensity of anticoagulation not defined | D |
Modification and/or treatment of risk factors for thrombotic events | D |
Table 22.3
Second line therapies (for life-threatening or catastrophic APS)
Therapy | Dose | Evidence level |
|---|---|---|
High-dose corticosteroids | Methylprednisolone 30 mg/kg (max 1 g) daily for 3 days, followed by prednisone 2 mg/kg/day | E |
Intravenous Immunoglobulin | 2 g/kg (max 70 g), no defined dosing schedule | E |
Plasmapheresis | n/a | E |
Cyclophosphamide | 750 mg/m2 per week for 4 weeks | E |
Rituximab | 375 mg/m2 per week for 4 weeks | E |
Anetoderma
Lauren B. McCaffery, MD and Heather A. Brandling-Bennett, MD
Clinical Features
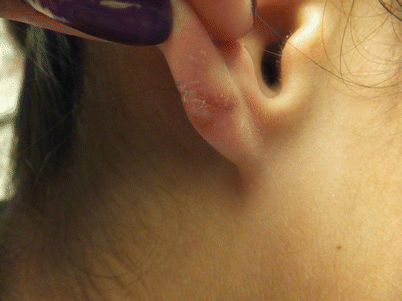
Anetoderma is a rare, benign condition of the skin characterized by localized patches of flaccid skin which can be macular, depressed (Fig. 22.1 and ), or appear as sac-like outpouchings of the skin. On examination, lesions tend to herniate with gentle pressure – an exam finding dubbed the “buttonhole sign.” Erythema and fine wrinkling of the overlying epidermis are variably present (Fig. 22.2) [11–14].
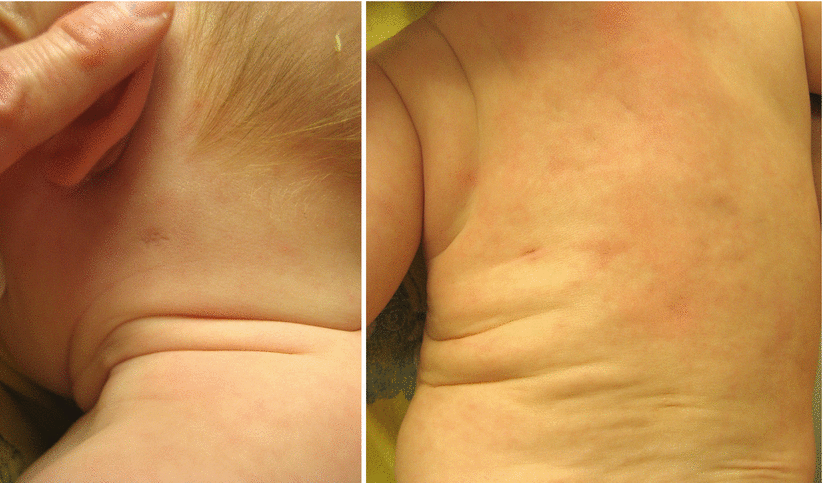
Fig. 22.1 and Fig. 22.2
An eight-month old infant was born with depressed, macular skin on the neck and back
Anetoderma can be divided into primary and secondary forms. Primary anetoderma arises in previously clinically normal skin, whereas secondary anetoderma arises at the site of skin previously or concomitantly involved with other dermatoses [14, 15]. Secondary anetoderma has been associated with numerous other conditions, most commonly varicella and acne, but also sarcoidosis, leprosy, human immunodeficiency virus (HIV), tuberculosis, and others [13, 14]. Historically, primary anetoderma was classified into two clinical categories: those associated with a preceding inflammatory stage (Jadassohn-Pelizzary type), and those without an inflammatory stage (Schweninger-Buzzi type). This distinction is primarily of historical interest, as the histologic findings and other clinical features are indistinguishable between these two types [14, 15].
Patients with primary anetoderma typically present with lesions ranging in number from few to over 100, with typical sites of involvement on the upper trunk and proximal extremities. Primary anetoderma can be associated with underlying systemic disease or immunologic abnormalities, most commonly the presence of antiphospholipid antibodies (aPL). Small case series have reported that the majority of patients with primary anetoderma have associated circulating aPL, and approximately half of these patients ultimately go on to develop full criteria for antiphospholipid antibody syndrome [13, 16, 17].
Anetoderma of prematurity is a distinct form of this disease, which occurs in premature infants receiving care in the neonatal intensive care unit (NICU). Dermatologic findings include the typical localized, flaccid patches of skin, but usually occur at sites of placement on monitoring leads. Accordingly, common sites of involvement include the periumbilical and subclavicular skin. This is thought to occur in immature skin either as a result of local hypoxia resulting from pressure at the site of monitoring leads, or from shearing stress from such leads. The incidence seems to be reduced when leads are placed ventrally on a supine neonate, and vice versa [18, 19].
Management Strategy
The diagnosis of anetoderma is based on typical clinical findings and supportive skin biopsy findings if the clinical diagnosis is in doubt. Anetoderma should be suspected in patients with the typical localized flaccid patches of skin. Histologic findings include focal loss of elastic fibers in the papillary dermis, often with an associated perivascular lymphohistiocytic inflammatory infiltrate [15, 17]. Even in patients with concomitant APS, the present of vascular microthrombi is uncommon [16]. Direct immunofluorescence (DIF) findings are not diagnostic, but can occasionally show overlapping features of a lupus band, with deposition of immunoreactants at the dermo-epidermal junction [16].
Given the high frequency of circulating aPL in patients with anetoderma, screening for aPL is warranted. Any patient with anetoderma and aPL should be monitored closely over time for development of other features of APS or other autoimmune conditions [13, 17].
Treatment for anetoderma has generally been unsatisfactory. Multiple treatments have been unsuccessfully attempted, including intralesional steroid injections, aspirin, phenytoin, dapsone, vitamin E, aminocaproic acid, and nicotinamide [15, 20]. Few case reports have shown success with penicillin or antimalarials [15]. Spontaneous resolution does not tend to occur [18]. For particularly cosmetically bothersome lesions, surgical excision can be performed [14]. Recently, treatment with destructive carbon dioxide (CO2) laser has been reported to lead to significant cosmetic improvement [21].
Investigations Recommended
For diagnostic |
Histology |
Biopsy of lesionsal skin, with elastic tissue stain |
Laboratory |
Antiphospholipid antibody panel |
Therapy | Dose | Evidence level |
|---|---|---|
First-line therapy | ||
Observation | N/A | N/A |
Second-line therapy | ||
Penicillin | 1800 IU daily for 3 weeks | E |
Hydroxychloroquine | 3–5 mg/kg/day divided BID, maximum 400 mg/day | E |
Surgical excision | N/A | E |
Destructive CO2 laser | N/A | E |
Atrophoderma of Pasini and Pierini
Regina-Celeste Ahmad, MD, PhD, Alex Fogel BS, and Joyce M.C. Teng, MD, PhD
Clinical Features
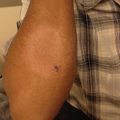
Atrophoderma of Pasini and Pierini (atrophoderma) is a benign, asymptomatic, idiopathic dermal atrophy disorder that develops as one or more round-to-ovoid, hyperpigmented, well-demarcated, non-indurated, depressed 1–12 cm patches lacking inflammation (Fig. 22.3) [22, 23]. The affected skin appears thin but feels normal, and the surrounding skin appears and feels normal [23]. Though this condition often arises in young adulthood, it is not uncommon in children, and congenital cases have been reported [24, 25].
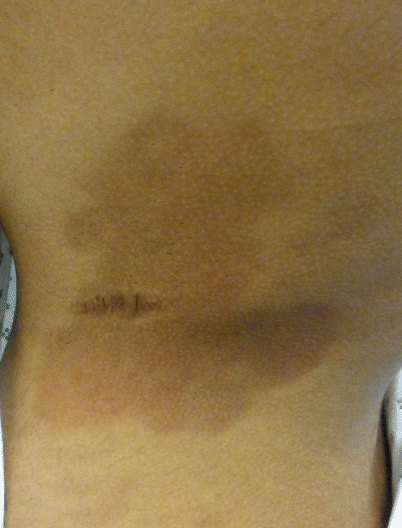
Fig. 22.3
Atrophoderma on the back of a sixteen year old girl. The onset was gradual over 2-3 year period
Though cases of atrophoderma with isolated lesions have been reported, the disease is typically characterized by multiple, bilaterally symmetric, discrete or confluent lesions [26]. Within a few weeks of arising, the lesions become hyperpigmented, and typically follow a course of slow enlargement over months to years before becoming quiescent [27]. As the lesions evolve, the initial hyperpigmentation may lighten, and the associated skin may become depressed, typically 1–8 mm below the level of the normal surrounding skin, resulting in the classic “cliff-drop” border pattern [28]. While these depressions are visible on side lighting, they are not usually palpable. Importantly, the normalcy of the surrounding skin differentiates atrophoderma from morphea [27].
The patches of atrophoderma typically arise during adolescence or early adulthood on the trunk, chest, arms and/or abdomen, while usually sparing the face, feet and hands [23]. The disease has been observed to co-exist with morphea and lichen sclerosis, and it is often included in differential diagnoses with these conditions, as well as with post-inflammatory hyperpigmentation and anetoderma [27]. Atrophoderma more commonly occurs in Caucasians than in people of darker skin color, in women than in men, and in Europeans more than North Americans [23, 25]. Some authors have suggested a relationship between Borrelia burgdorfi infection and disease development, but the evidence on this relationship is not definitive [29].
Management Strategy
Atrophoderma is a benign and asymptomatic disorder, with no affect on life expectancy or overall health. The disease typically follows a protracted course with symptoms persisting 10–20 years, though spontaneously self-resolution is not uncommon.
There are currently no accepted standard therapies, due to the rarity of the condition and the absence of good clinical trials. Patient reassurance is thus of primary importance. Patients typically seek treatment to rule out more serious conditions and to alleviate the undesirable appearance.
Sun exposure is believed to darken hyperpigmented lesions in atrophoderma [30]. Photoprotection is therefore recommended to reduce further discoloration of the skin.
Specific Investigations
Side lighting during physical exam
Skin Biopsy
Side lighting may be used to observe the characteristic “cliff-drop” borders of atrophoderma. Single lesions have been described as “inverted plateaus” and multiple lesions have been described as having a “Swiss cheese” appearance.
While atrophoderma is a diagnosis of exclusion, skin biopsy may be useful to exclude other conditions. Wedge or elliptical excisions are often recommended over punch biopsy for atrophic conditions to minimize sampling bias. A rim of normal skin should be included in the biopsy sample [28]. Observed histopathological changes in atrophoderma are minimal and non-diagnostic, though specimens often display decreased dermal thickness when compared to normal skin specimens [29].
First-Line Therapies
Patient counseling and reassurance
Patients should be reassured that atrophoderma is a benign condition without additional associated risk to overall health or life expectancy. Patients should be reassured that the lesions are not infectious, and physicians should palpate the lesions of atrophoderma without gloves. Patient concerns, such as cosmetic appearance, should be understood and used to guide management.
Second-Line Therapies
The following therapies are all level five evidence from adult studies:
Topical corticosteroids
Antibiotics
Antimalarials
Laser therapy
While no treatment has been shown to be consistently effective for atrophoderma, response to topical corticosteroids, antibiotics and antimalarials have been reported in sporadic cases [23, 31, 32]. There is no agreed-upon dosing regimen. Additionally, the efficacy of antimicrobial therapy in atrophoderma has been primarily observed during treatment of infectious disease [32]. It is therefore unclear whether the drug itself induced improvement in the lesions of atrophoderma, or whether clearance of the underlying infectious organism resulted in improvement of the lesions [29] More study in this area is needed.
Surgical treatment has not been shown to be effective in restoring the appearance of the affected skin. Laser therapy to reduce hyperpigmentation has been described in one case using a Q-switched alexandrite laser (755 nm) [30]. This therapy induces selective photothermolysis of pigment in the epidermis. However, further research is needed to confirm these findings.
Degos Disease (Malignant Atrophic Papulosis)
Kate Khorsand, MD, and Heather Brandling-Bennett, MD
Clinical Features
Degos disease, also known as malignant atrophic papulosis, is a rare disorder thought to be caused by a thrombotic vasculopathy. There are two forms of the condition, with one form limited to skin findings and the other with systemic involvement. The systemic form carries a vastly worse prognosis, with rapidly progressive disease that is nearly universally fatal within several years. The skin-limited form can progress into systemic disease months to years later [33]. However, it is possible that the incidence of skin-limited Degos disease is underreported given its benign course. The disease is very rare in children and infants [34, 35].
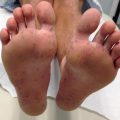
Degos disease presents with the pathognomonic finding of erythematous papules which develop a pearly, atrophic, porcelain white center often with a telangiectatic rim (Figs. 22.4 and 22.5) [36]. The palms, soles, face and scalp are rarely involved [37]. The gastrointestinal tract and central nervous system are most commonly affected in the systemic form, but other organs may be involved including the pericardium, lungs, eyes, and bladder. Morbidity and mortality is usually caused by sepsis or hemorrhage from bowel or brain infarction. Degos-like lesions may be seen in patients with systemic lupus erythematosus, antiphospholipid antibodies, or dermatomyositis.
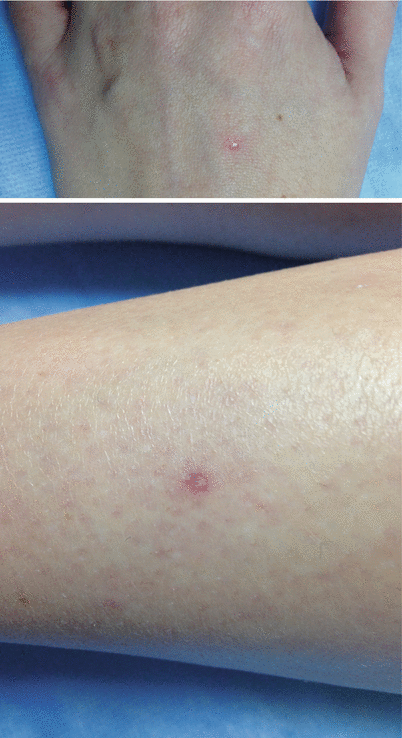
Fig. 22.4 and Fig. 22.5
A seventeen-year old Caucasian female presented with atrophic macules on the dorsal hand and lower extremities. The lesions had white centers and erythematous rims
The pathophysiology of this disease is unclear, with several hypotheses in existence. The three leading hypotheses at this time favor either inflammation of the vessels, coagulopathy, or primary dysfunction of endothelial cells as the trigger for development of the disease [37].
Management Strategies
Skin lesions suspicious for Degos disease are typically biopsied, and histopathology reveals wedge-shaped dermal necrosis with an overlying atrophic epidermis and hyperkeratosis. There are no diagnostic laboratory markers used, however, coagulation parameters are often abnormal [37]. Patients should be carefully evaluated for any signs of systems of systemic disease, with additional testing performed as indicated below [33]. Patients with skin-limited Degos disease should be followed closely for several years. Unfortunately there is no uniformly effective therapy, and treatment does not seem to prevent progression of the disease from the skin-limited form to the systemic form [37]. The skin-limited form may not be treated, as it is considered a benign process, and immunosuppressive agents can worsen cutaneous lesions.
Investigations Recommended
For diagnosis |
Skin biopsy |
Fecal occult blood test |
Opthalmologic referral and ocular fundus examination |
If systemic disease suspected: colonoscopy, brain MRI, echocardiogram |
First Line Therapies
Patients with skin-limited disease may be conservatively managed by close monitoring. The first line therapeutic approach for a patient with systemic Degos disease should optimize perfusion and minimize thrombus formation [37]
Therapy | Dose | Evidence |
|---|---|---|
Optimize perfusion, minimize thrombus formation | ||
Aspirin | 325 mg PO dailya | Eb |
Pentoxyfylline | 400 mg PO three times dailya | Eb |
Dipyridamole | 75–100 mg PO four times dailya | Eb |
Ticlopidine | 250 mg PO twice dailya | Eb |
Vasodilation, inhibition of platelet aggregation | ||
Treprostinil | Dose variable | Eb |
Immunosuppression | ||
Cyclosporine | 3.5–5 mg/kg/day | Eb |
Azathioprine | Start 1–3 mg/kg/day with max of 250 mg daily | Eb |
Cyclophosphamide | 500–1000 mg/m2 IV monthly | Eb |
Second Line Therapy
Several case reports have seen dramatic improvement with eculizumab, a humanized monoclonal antibody acting as a terminal complement inhibitor. This is thought to counteract the complement activation and increased endothelial cell apoptosis leading to thrombotic complications in systemic Degos disease. Unfortunately, this treatment was eventually unable to halt the progression of systemic disease [37, 38].
Dermatomyositis
Nina Washington, MD and Joyce M.C. Teng, MD, PhD
Clinical Features
Juvenile dermatomyositis (JDM) is an autoimmune inflammatory vasculopathy. It classically manifests as symmetric proximal muscle weakness and rash. At diagnosis, constitutional symptoms including fever, fatigue, and weight loss are common [39, 40].
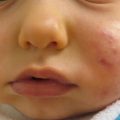
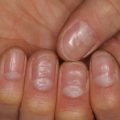
The pathognomonic rash of JDM is the Gottron’s Papule – a flat-topped pink to violaceous papule located on the metacarpophalangeal and interphalangeal joints (Fig. 22.6). Classic rashes associated with JDM also include: (1) Heliotrope rash – a violaceous rash of the eyelids often accompanied by periorbital edema and/or erythema; (2) V sign – an erythematous macular rash of the sun-exposed areas of the neck and chest; and (3) Shawl sign – an erythematous macular rash of the back of the neck and shoulders. Patients may also have a malar rash similar to that seen in lupus patients, periungual erythema and telangiectasias of the nail bed capillaries, ulcerations (Fig. 22.7), and erythematous rashes of the extensor surfaces of the knees and elbows (Gottron Sign) [41].
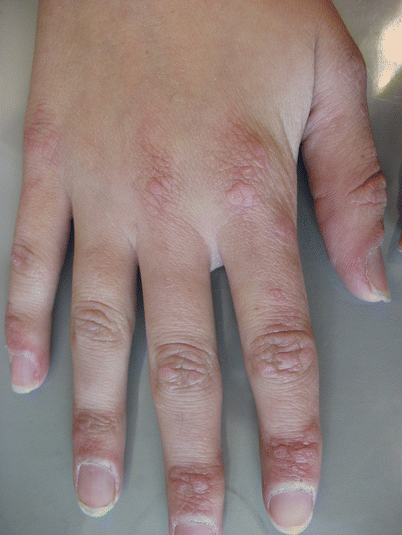
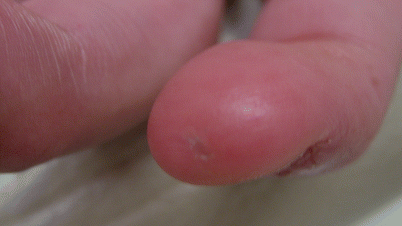
Fig. 22.6
Gottron’s papules on the dorsal hand of a teenager with DM. The lesions clustered over the metacarpophalangeal (MCP), proximal interphalangeal (PIP) and distal interphalangeal (DIP) joints
Fig. 22.7
Painful skin ulceration noted on the finger of a teenager with DM
Muscle weakness in JDM is insidious in nature. Patients may have trouble combing or brushing their hair, getting dressed, climbing stairs, or sitting in a chair. Respiratory distress may arise if the diaphragm is affected. Patients may experience dysphagia or hoarseness if the pharyngeal muscles are affected.
Management Strategies
To date, there have been no randomized, controlled clinical trials (RCTs) solely investigating the treatment of juvenile dermatomyositis; in part due to the rarity and heterogeneity of the disease as well as ethical concerns regarding participation of children in RCTs. Currently, treatment for JDM is divided into topical and systemic therapy, based on consensus guidelines [42, 43] Standard treatment includes corticosteroids and immunosuppressant agents. Methotrexate is overwhelmingly the immunosuppressant of choice amongst providers. Once systemic inflammation is optimally controlled, physical therapy serves to restore muscle strength and conditioning.
Specific Investigations Recommended
In 1975, Bohan and Peter developed criteria for the diagnosis of dermatomyositis [39–41]. Clinical features consistent with a diagnosis included: (1) characteristic skin findings (i.e., Gottron’s papules, heliotrope rash, Shawl sign, and malar rash); (2) proximal muscle weakness; (3) elevated muscle enzyme levels; (4) a myopathic pattern on EMG; and (5) evidence of an endomysial, mononuclear inflammatory infiltrate on muscle biopsy. A summary of recommended diagnostic investigations is detailed here.
Diagnostic investigations | |
|---|---|
Laboratory | CK, LDH, aldolase, ALT, AST, Anti-Nuclear Antibody, Anti-Jo-1, Anti-Ro/SSA |
Imaging | MRI |
Functional testing | Nerve Conduction Study (NCS), electromyography |
Histology | Muscle biopsy |
First Line Therapies
In both consensus publications by the Childhood Arthritis and Rheumatology Research Alliance (CARRA), pediatric rheumatologists agreed that high-dose corticosteroids and methotrexate should be used concurrently as first-line therapy in patients with JDM [42, 43].
Therapy | Dose | Evidence level |
|---|---|---|
Topical therapy | ||
Topical corticosteroids (e.g., desonide, clobetasol) | Apply to affected areas daily to twice daily | D |
Topical calcineurin inhibitors (e.g., tacrolimus, pimecrolimus) | Apply to affected areas twice daily | D |
Systemic therapy | ||
Corticosteroids | Intravenous methylprednisolone – 30 mg/kg/day (max 1 g) for 3 days | D |
Transition to oral prednisone at 2 mg/kg/day for 4 weeks | ||
Subsequently tapered by 20 % by treating physician | ||
Recommended duration 1 year | ||
Methotrexate | The lesser of 15 mg/m2 or 1 mg/kg (maximum 40 mg) once weekly | D |
Subcutaneous route recommended | ||
Folic acid supplementation required (1 mg po daily) | ||
Second Line Therapies
IVIg and Hydroxycholorquine are also effective in the treatment of JDM skin disease [44]. In some protocols, they may be used as first line therapy.
Therapy | Dose | Evidence level |
|---|---|---|
Intravenous immunoglobulin (IVIg) | 2 g/kg (maximum 70 g) | C |
Recommend dosing schedules: | ||
Every 2 weeks × 3, then monthly | ||
Over 2–5 days then 0.4–2 mg/kg/month | ||
Hydroxychloroquine (Plaquenil) | 6–7 mg/kg/day divided bid (maximum 400 mg daily) | E |
Third Line Therapies
Third line therapies are used for refractory disease or organ involvement such as interstitial lung disease (ILD).
Therapy | Dose | Evidence level |
|---|---|---|
Cyclosporin A | 3.5–5 mg/kg/day | Ba |
Azathioprine (imuran) | 1–3 mg/kg/day with max of 150 mg daily | D |
Mycophenolate mofetil (CellCept) | 20 mg/kg divided twice daily or, | |
800–1350 mg/m2/day | ||
Total dose of 1–3 g/day | ||
Tacrolimus (prograf) | 0.1–0.2 mg/kg/day divided twice daily | D |
Cyclophosphamide (cytoxan) | 500–1000 mg/m2 administered monthly × 6 months | D |
Rituximab (rituxan) | BSA ≤1.5 m2, 575 mg/m2 at week 0 and week 2 | A |
BSA >1.5 m2, 750 mg/m2 (up to 1 g/infusion) at weeks 0 and 2 |
Non-pharmacologic Therapies
Physical/occupational therapy |
Calcium and vitamin D |
Sunscreen (minimum SPF 30) |
Photoprotective clothing |
Eosinophilic Fasciitis
Shelley Yang, MD, Regina-Celeste Ahmad, MD, and Heather A. Brandling-Bennett, MD
Clinical Features
Eosinophilic fasciitis (EF), also known as Shulman’s syndrome, is a rare idiopathic fibrosing disorder characterized by inflammation of the fascia resulting in edema and induration of the skin.
EF affects patients of all ages, but is more common in adults with a mean age of onset between 40 and 50. Only a few dozen cases have been reported in the pediatric population. In children, there appears to be a female predominance. Strenuous physical exertion has been reported as a risk factor in multiple case series. Other proposed triggers include trauma, drugs, infection, solid neoplasms, and hematologic disorders.
Presentation is characterized by rapid onset of painful edema and erythema, evolving into induration and fibrosis of the subcutaneous fascia. Distribution is usually symmetrical involvement of the extremities and sometimes the trunk, sparing the face. In children, the hands are frequently affected. The affected skin acquires a bound down, peau d’orange quality. The “groove sign” has been described in reference to an indentation along the course of superficial veins, and is caused by retraction of the subcutaneous tissue. Some children initially diagnosed with EF have evolved into generalized or localized morphea, suggesting EF may be a variant of morphea.
Patients may develop carpal tunnel syndrome, arthritis, and paresthesias. Compared to the adult population, arthritis is less common in children with EF, and there is a lower incidence of hematological abnormalities such as thrombocytopenia, as well as hematologic and solid organ malignancies. Renal involvement, especially IgA nephropathy, has been described in children.
Characteristic laboratory abnormalities include peripheral eosinophilia, hypergammaglobulinemia, and an elevated sedimentation rate.
Management Strategies
Early initiation of systemic prednisone is the mainstay of treatment. Patients usually show a good clinical response with gradual improvement of induration and resolution of laboratory abnormalities. There is also potential for spontaneous remission which can complicate evaluation of treatment response.
Younger onset age (<7 years old) and greater disease severity have been associated with a higher risk of progression and fibrosis refractory to treatment. Therefore, early physical therapy may be an important adjunct to treatment.
Investigations Recommended
There are no published consensus diagnostic criteria for EF. Defining features based on Shulman’s original description in 1974 include rapid onset symmetric skin induration along with transient eosinophilia, hypergammaglobulinemia, and marked favorable response to systemic corticosteroids in the absence of Raynaud’s phenomenon or any internal manifestations of systemic sclerosis. A deep excisional biopsy down to fascia is recommended for histological diagnosis. Histology reveals fibrosis of the deep fascia and subcutis with a chronic inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells. Eosinophils may be found in the lower subcutis and fascial layers, but can be only focal and transient, and are not always observed or necessary for diagnosis. Some authors believe a clinical diagnosis based on appearance, labs, and MRI findings are sufficient, without absolute need for surgical biopsy. MRI can be used to localize the optimal anatomic location for a biopsy and monitor response to therapy. Characteristic findings on MRI include superficial fascial thickening, abnormal signal intensity, and contrast enhancement.
Laboratory evaluation typically includes testing for complete blood count with differential and peripheral smear, total immunoglobulin levels, inflammatory markers, muscle enzymes, thyroid studies and autoantibodies. A positive ANA has been found in less than 25 % of patients. Peripheral eosinophilia is present in the majority of patients but is not required for diagnosis and is not an indicator of disease activity. Aldolase, a muscle enzyme, may be a useful measure of disease activity.
First Line Therapies
EF is managed initially with high dose corticosteroids Evidence level D [47–49]. Prednisone or prednisolone is typically given at doses of approximately 0.5–1.0 mg/kg/day. Methylprednisolone pulses (500–1000 mg daily for three consecutive days) Evidence level D 1 [49] before prednisone treatment has been associated with better outcomes.
Second Line Therapies
Methotrexate at low doses (15–25 mg once weekly) Evidence level D [49] is the preferred second-line treatment either alone or in combination with systemic corticosteroids, especially in patients with morphea-like skin lesions or unsatisfactory responses to systemic corticosteroids. In adults, good responses were found to hydroxychloroquine (200–400 mg daily) Evidence level D 1 [49] in combination with prednisone.
Third Line Therapies
Third line treatments described in pediatric patients with poor response to systemic corticosteroids include d-penicillamine, mycophenolate mofetil, and infliximab Evidence level E [47, 50, 51]. In adults, dapsone, cyclosporine, azathioprine, intravenous immunoglobulin, rituximab, UVA1 phototherapy, and extracorporeal photochemotherapy have been used Evidence level E 1 [52–58].
Juvenile Idiopathic Arthritis
Joy Mombourquette, MD, and Joyce M.C. Teng, MD, PhD
Clinical Features
Juvenile idiopathic arthritis, or JIA, is a general term encompassing several categories of chronic arthritis with onset before the age of 16 years. The instigating cause of JIA is unknown, but the inflammation of the disease is caused by dysfunction of the immune system. Current research suggests that most types of JIA are predominately caused by autoimmunity (dysfunction of the adaptive immune system), with the exception of Systemic JIA, which is thought to be predominately autoinflammatory (dysfunction of the innate immune system).
According to the International League of Associations for Rheumatology (ILAR) criteria, JIA is divided into seven different categories: (1) Oligoarticular JIA, (2) RF negative Polyarticular JIA, (3) RF positive Polyarticular JIA, (4) Systemic JIA, (5) Juvenile Psoriatic Arthritis, (6) Enthesitis-Related Arthritis, and (7) Undifferentiated JIA. Undifferentiated JIA includes chronic arthritis that fulfills criteria for no category or overlaps two or more other categories [59].
Oligoarticular JIA is defined as arthritis in four or fewer joints during the first 6 months of disease. Oligoarticular JIA has a peak onset in the preschool years and affects females more than males. These patients are at higher risk than other categories of JIA for developing chronic uveitis, an inflammatory eye disease that is often insidious in onset.
Polyarticular JIA is arthritis in five or more joints. It also affects females more than males, with two peaks of onset in the preschool years and the later childhood/adolescence years. Polyarticular JIA is divided under the ILAR criteria into rheumatoid factor (RF) positive and negative disease. The arthritis in patients with RF positive disease has a poorer prognosis and tends to occur in older children and adolescents [60].
Systemic JIA is characterized by the classic triad of fever, rash, and arthritis. The fever needs to be present for at least 2 weeks and be quotidian (daily). The classic rash is a salmon-colored, evanescent rash that occurs most often on the trunk and proximal extremities, and usually appears during the times of fevers, with resolution or fading between fevers. The rash can sometimes be induced by heat or by the Koebner phenomenon, can occasionally be pruritic, and can precede the onset of arthritis by days to years [61]. The arthritis can involve any number of joints, but is typically polyarticular, and needs to be present for 6 weeks to meet diagnostic criteria. Patients often feel ill at times of fever, with return to baseline or near baseline between episodes. Other diagnostic criteria that can be present include hepatosplenomegaly, lymphadenopathy, and serositis.
Enthesitis-related arthritis, or ERA, is a member of the Sponyloarthropathy arthritis family, and involves inflammation of the entheses sites. Common areas of involvement include the sacroiliac joints, the insertion site of the Achilles, quadriceps, and patellar tendons, and the insertion sites of the plantar fascia. The spine can also become involved. This type of arthritis primarily occurs in older children and adolescents, and is more common in males than females. Patients with ERA are more likely to develop an acute type of uveitis that presents suddenly with a painful red eye.
Juvenile psoriatic arthritis has two distinct peaks of onset. The first peak is in the preschool years, and these patients tend to present with polyarticular arthritis and dactylitis. The second peak is in older children and adolescents. These patients tend to have more features of enthesitis and axial joint involvement [62]. Psoriasis does not need to be present to diagnosis psoriatic arthritis, and patients can develop skin disease years after their arthritis features first appear. Additional diagnostic criteria include nail pits, onycholysis, or history of psoriasis in a first-degree relative [59].
Management Strategies
Investigation Recommended
Obtain detailed history about fever, rash and joint symptoms Radioimaging studies i.e. x-ray or MRI if indicated
Specific Therapies
Treatment of JIA is largely dependent on the type of JIA. The paragraphs and table below provide a brief, concise overview of different medications used in the treatment of JIA. It is strongly recommended that therapy of patients with JIA be administered in conjunction with the recommendations of, and under the supervision of, a pediatric rheumatologist or an adult rheumatologist with experience in the treatment of JIA. Several recommended treatment pathways for the treatment of JIA are available from the American College of Rheumatology and the Childhood Arthritis and Rheumatology Research Alliance (CARRA).
Non-steroidal anti-inflammatory drugs (NSAIDs) are often part of first-line treatment for JIA, but are usually used in combination with a disease-modifying antirheumatic drug (DMARD) and/or biologic medication. There are numerous NSAIDs used in pediatric patients, with varying dose schedules, side effects, and availability of liquid formulations. The most commonly used NSAIDs include ibuprofen, naproxen, meloxicam, diclofenac, indomethacin, and sulindac. In order to achieve anti-inflammatory effects, doses need to be administered on an around-the-clock schedule, and are often administered at higher doses than those used for analgesic effects. For further information on specific NSAID dosing and administration, the author recommends referencing additional resources.
Systemic corticosteroids are generally used sparingly in the treatment of JIA, due to their significant toxicity profile. Doses vary depending on the type of corticosteroid, the level of immunosuppression intended, the route of administration, and the planned duration of treatment. Systemic corticosteroids are recommended as an option for first-line treatment of systemic JIA with or without the use of other medications, but are otherwise not recommended as first-line therapy in other forms of JIA [63]. They can be used acutely during times of disease flare to quickly control inflammation, or to bridge therapy while starting or transitioning DMARDs and biologic medications.
Side effects of systemic corticosteroids include, but are not limited to, increased appetite and weight gain, striae, systemic hypertension, intraocular hypertension, hyperglycemia, mood changes, cataracts, glaucoma, acne, myalgias, growth suppression, osteoporosis, immunosuppression, impaired wound healing, and avascular necrosis. Intra-articular corticosteroid injections are used as treatment in all forms of JIA, and can be used alone or in conjunction with other medications. Intra-articular corticosteroids are useful for providing anti-inflammatory treatment to individual affected joints while avoiding many of the systemic side effects seen with the use of systemic corticosteroids. Injections can be given up to every 3 months as needed for active arthritis, but remission in a joint after injection can last for over 2 years [64]. Side effects of intra-articular corticosteroid injections include skin atrophy and/or hypopigmentation at the injection site, infection, bleeding, or tendon rupture.
Many JIA patients require initiation of a DMARD and/or a biologic at the time of diagnosis or soon after diagnosis, depending on their type of JIA and on their presentation (duration of symptoms, progression of symptoms, signs of articular damage on imaging). The decision of a particular DMARD and/or biologic medication used is based on the type JIA, patient characteristics (ex. presence of uveitis), and features of disease (ex. pattern of involved joints, presence of joint erosions on imaging).
Medication family | Medication | Dose | Common and serious side effects | Lab monitoring | Indications | Level of evidence |
|---|---|---|---|---|---|---|
Disease-modifying antirheumatic drugs (DMARDs) | 10–20 mg/m2/week by mouth or subcutaneous injection (max dose 25 mg); subcutaneous injections are recommended for higher doses | Nausea, vomiting, anemia, hepatotoxicity, nephrotoxicity, bone marrow suppression, teratogenicity, immunosuppression | Complete blood count with differential, renal and liver function testing initially and every 1–2 months, spaced out to every 3–4 months once on stable doses | Oligoarticular JIA | A [66] | |
Polyarticular JIA | ||||||
Systemic JIA | ||||||
ERA | ||||||
Juvenile psoriatic arthritis | ||||||
All doses given orally: | Rash, nausea, diarrhea, headaches hepatotoxicity, bone marrow suppression, teratogenicity, immunosuppression | Complete blood count with differential, liver function testing and albumin initially and every month for the first 6 months, spaced out to every 6–8 weeks thereafter | Often used interchangeably in patients who cannot tolerate methotrexate | A (studied in patients with polyarticular JIA) [69] | ||
<20 kg: loading dose of 100 mg × 1 day, then 10 mg every other day | ||||||
20–40 kg: loading dose of 100 mg × 2 days, then 10 mg daily | ||||||
>40 kg: loading dose of 100 mg × 3 days, then 20 mg daily | ||||||
15–25 mg/kg by mouth –twice daily (maximum daily dose 2 g) | Nausea, vomiting, dyspepsia, anemia/bone marrow toxicity, rash, reversible oligospermia | Complete blood count with differential, renal and liver function testing prior to starting therapy, every 2 weeks for the first 3 months, every 4 weeks for the next 3 months, then every 12 weeks while on stable doses | Oligoarticular JIA | A (Oligoarticular and polyarticular JIA) [70] | ||
Polyarticular JIA | A* (Spondyloarthritis with only peripheral articular disease) [71] | |||||
ERA | ||||||
Juvenile psoriatic arthritis | ||||||
*Due to side effects, recommend starting at 5 mg/kg twice daily and increasing weekly by 10 mg/kg/day to goal dose | Hypersensitivity reactions: Stevens-Johnson Syndrome and Drug Rash with Eosinophilia and Systemic Symptoms Syndrome (DRESS) | |||||
Avoid in patients with allergy to sulfa drugs, with Glucose-6-Phosphate Dehydrogenase deficiency, and porphyria | ||||||
Cyclosporine [73] | 2–5 mg/kg/day by mouth, usually divided twice daily | Nausea, vomiting, hypertrichosis, gingival hyperplasia, mucosal lesions, hypertension, renal toxicity, hepatic toxicity, immunosuppression | Blood pressures, complete blood counts with differential, renal and liver function studies, and trough drug levels periodically | Oligoarticular JIA | D [75] | |
Polyarticular JIA | ||||||
Systemic JIA | ||||||
Systemic JIA (dose from case studies): 0.075–0.11 mg/kg/day by mouth, divided twice daily | Rash, nausea, vomiting, diarrhea, constipation, headaches, paresthesias, hypertension, renal toxicity, hepatic toxicity, hypertriglyceridemia, hyperglycemia, diabetes mellitus, electrolyte abnormalities, immunosuppression | Blood pressures, complete blood counts with differential, renal and liver function studies, electrolytes including magnesium and glucose levels, and trough drug levels periodically | Systemic JIA | |||
Polyarticular JIA | ||||||
(Not studied in Oligoarticular JIA, but can be considered in place of cyclosporine) | ||||||
Tumor necrosis factor (TNF) alpha inhibitors | 5–10 mg/kg/dose intravenously; second dose given 2 weeks after the first dose, with subsequent dosing every 4 weeks | Increased risk for the development of opportunistic infections, including invasive fungal infections. Reactivation of latent tuberculosis. Infusion reactions, formation of human anti-chimeric antibodies (HACA), induction of additional autoimmunity, development or worsening of psoriasis, increased risk of malignancy, new-onset or exacerbation of demyelinating diseases and optic neuritis, worsening heart failure in patients with a history of heart failure or decreased left ventricular function | Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis | Oligoarticular JIA | B (Polyarticular disease in conjunction with methotrexate) [80] | |
Polyarticular JIA | ||||||
Systemic JIA | ||||||
ERA | ||||||
Complete blood counts with differential, renal and liver function testing every 4–12 weeks | Juvenile Psoriatic Arthritis | |||||
FDA approved for patients ages 2 years and older: 0.4 mg/kg/dose subcutaneously twice weekly (maximum dose 25 mg). Alternative dosing 0.8 mg/kg once weekly (maximum dose 50 mg) | Increased risk for the development of opportunistic infections, including invasive fungal infections. Reactivation of latent tuberculosis. Injection site reactions, induction of additional autoimmunity, development or worsening of psoriasis, increased risk of malignancy, new-onset or exacerbation of demyelinating diseases and optic neuritis, worsening heart failure in patients with a history of heart failure or decreased left ventricular function | Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis | Oligoarticular JIA | A (Polyarticular JIA) [84] | ||
Complete blood counts with differential, renal and liver function testing every 4–12 weeks | Polyarticular JIA | |||||
Systemic JIA | ||||||
ERA | ||||||
Juvenile Psoriatic Arthritis | ||||||
FDA approved for ages 2 years and older: | Increased risk for the development of opportunistic infections, including invasive fungal infections. Reactivation of latent tuberculosis. Injection site reactions, induction of additional autoimmunity, development or worsening of psoriasis, increased risk of malignancy, new-onset or exacerbation of demyelinating diseases and optic neuritis, worsening heart failure in patients with a history of heart failure or decreased left ventricular function | Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis | Oligoarticular JIA | A (Polyarticular JIA) [88] | ||
10–15 kg: 10 mg subcutaneously every other week | Complete blood counts with differential, renal and liver function testing every 4–12 weeks | Polyarticular JIA | ||||
15–30 kg: 20 mg subcutaneously every other week | Systemic JIA | |||||
>30 kg: 40 mg subcutaneously every other week | ERA | |||||
Juvenile psoriatic arthritis | ||||||
Cytotoxic T lymphocyte antigen immunoglobulin (CTLA-4Ig) | Intravenous formulation FDA approved for patients 6 years and older: | Increased risk for the development of opportunistic infections and reactivation of latent tuberculosis and Hepatitis B. Infusion reactions. Headaches, nausea, rash, and hypertension | Testing for Hepatitis B prior to starting therapy. Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis | Polyarticular JIA | A (Extended oligoarticular JIA, polyarticular JIA, systemic JIA without systemic features) [93] | |
Weight <75 kg: 10 mg/kg/dose | Extended oligoarticular JIA | |||||
75 kg–100 kg: 750 mg/dose | Systemic JIA | |||||
>100 kg: 1000 mg/dose | ||||||
Dosing: Weeks 0, 2, and 4, then every 4 weeks | ||||||
Anti-CD20 antibody | Dosing used in pediatric studies: | Immunosuppression, reactivation of Hepatitis B infection, infusion reactions, hypogammaglobulinemia. Increased risk of the development of Progressive Multifocal Leukoencephalopathy | Testing for Hepatitis B prior to starting therapy. B cell levels before and 1 month after dosing. Qualitative immunoglobulins every 3 months. Complete blood counts with differentials at regular intervals | Oligoarticular JIA | B (Oligoarticular JIA, polyarticular JIA, and systemic JIA) [96] | |
375 mg/m2 IV once weekly for 2–4 doses | Polyarticular JIA | |||||
OR | Systemic JIA | |||||
500 or 750 mg/m2 IV per dose given 2 weeks apart, for 2 doses | ||||||
Maximum dose of 1000 mg/dose | ||||||
*In pediatric studies, repeat dosing was given ≥24 weeks after the first dose, at the time of reappearance of symptoms | ||||||
IL-1 inhibitors | Typical dosing used in Systemic JIA: 1–2 mg/kg daily, increasing to 4 mg/kg/day as needed for continued uncontrolled disease | Immunosuppression, reactivation of latent tuberculosis, injection site reactions, transaminitis and hepatitis, neutropenia | Testing for latent tuberculosis prior to starting therapy, annually, and with any high risk exposures to tuberculosis. Complete blood count with differential, LDH, and serum creatinine at baseline, weeks 1 and 2, months 1, 2, 6, and 9, and with dose changes | Polyarticular JIA | ||
Dose studied in Polyarticular JIA: 1 mg/kg/day | Systemic JIA | |||||
Maximum adult dose: 100 mg/day | ||||||
– | FDA approved for children ≥2 years of age and weight ≥7.5 kg: 4 mg/kg/dose every 4 weeks (maximum dose 300 mg) | Immunosuppression, reactivation of latent tuberculosis, injection site reactions, transaminitis and hepatitis, neutropenia and other cytopenias | Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis. Complete blood counts with differentials periodically. Consider obtaining liver function testing periodically | Systemic JIA. | A (Systemic JIA) [102] | |
– | 4.4 mg/kg (maximum dose 320 mg) × 1, then 2.2 mg/kg (maximum dose 160 mg) once weekly | Immunosuppression, reactivation of latent tuberculosis, injection site reactions, and hyperlipidemia | Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis. Complete blood counts with differentials periodically. Serum lipid levels every 2–3 months after starting therapy and then periodically | Systemic JIA | B (Systemic JIA) [104] | |
IL-6 receptor inhibitor | Intravenous dosing FDA approved for children 2 years of age and older: | Immunosuppression, reactivation of latent tuberculosis, infusion reactions, hyperlipidemia, neutropenia, thrombocytopenia, and transaminitis | Testing for latent tuberculosis prior to starting treatment and with any high risk exposures to tuberculosis. Complete blood count with differential and liver function testing with second infusion and every 1–2 infusions thereafter. Serum lipid levels should be obtained every 4–8 weeks initially, then every 6 months | Polyarticular JIA | ||
Weight <10 kg: 10 mg/kg/dose | Systemic JIA | |||||
Weight ≥30 kg: 8 mg/kg/dose | ||||||
Maximum dose: 800 mg | ||||||
Dosing for Systemic JIA: Every 2 weeks | ||||||
Dosing for Polyarticular JIA: Every 4 weeks |
Lupus Erythematosus
Rebecca Kunder, MD and Joyce M.C. Teng, MD, PhD
Clinical Features
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multi-organ dysfunction caused by auto-antibody production. Antinuclear antibodies (ANA) are present in nearly all patients with SLE, but also present approximately 10 % of healthy children. Antibodies that are more specific but less sensitive for SLE include antibodies to double-stranded DNA (dsDNA) and particular RNA-associated protein complexes (Smith and U1RNP). About 20 % of patients with SLE present during childhood, mostly during the teenage years, and there is a female predominance. Presenting symptoms can be constitutional such as fever, fatigue, and weight loss. End-organ damage may be present at diagnosis as well. There are two main sets of criteria for SLE diagnosis [108, 109], each of which rely on a combination of immunologic and clinical criteria. The main distinction of the more recent Systemic Lupus International Collaborating Clinics (SLICC) criteria as opposed to the American College of Rheumatology (ACR) criteria is that biopsy-proven lupus nephritis can be used to make the diagnosis under the SLICC guidelines (Tables 22.4 and 22.5). The clinical manifestations of SLE are protean, but commonly include hematologic, mucocutaneous, musculoskeletal, neurologic and renal pathology. Focusing on the dermatologic manifestations, up to 85 % of patients with SLE have a malar rash or other cutaneous findings at some point. In fact, SLE was so named because the facial rash was thought to resemble the bite of a wolf (“lupus” in Latin). The malar rash is characterized as an area of fixed erythema predominantly over malar eminences which spares the nasolabial folds and often appear to be “butterfly” shaped. It commonly involves the chin and can be either raised or flat. Cutaneous eruption in SLE is often in photo-exposed area, therefore can be used as a diagnostic criterion. Of note, discoid lesions are much less common in SLE than they are in isolated discoid lupus erythematosus (Figs. 22.8 and 22.9). The cutaneous manifestations of SLE, which can be seen in a variety of autoimmune diseases with a vasculitic component, are often polymorphic. In addition to erythematous patches and plaques in photosensitive area ulcers (both on the skin and the oral mucosa), purpura, as well as color change of the extremities (Raynaud’s phenomenon), are also frequent findings in patients with SLE. Importantly, patients who initially have cutaneous findings only can later develop systemic disease; approximately 25 % of SLE patients initially come to medical attention due to rash.