Key points
- •
Atopic dermatitis (AD) is a complex disease with various types of leukocytes involved in its pathology.
- •
Multiple subsets of CD4 + helper T cells, which are sensitized, activated, and differentiated by antigen presenting cells (i.e., Langerhans cells and dendritic cells) in damaged skin and Staphylococcus aureus infection, lead to acute and chronic inflammation in AD.
- •
Mast cells, granulocytes (basophils and eosinophils), and group 2 innate lymphoid cells (ILC2) contribute to the onset and development of skin inflammation.
- •
Advanced knowledge about cellular factors in AD pathology may lead to novel strategies for treatment of such disease.
Introduction
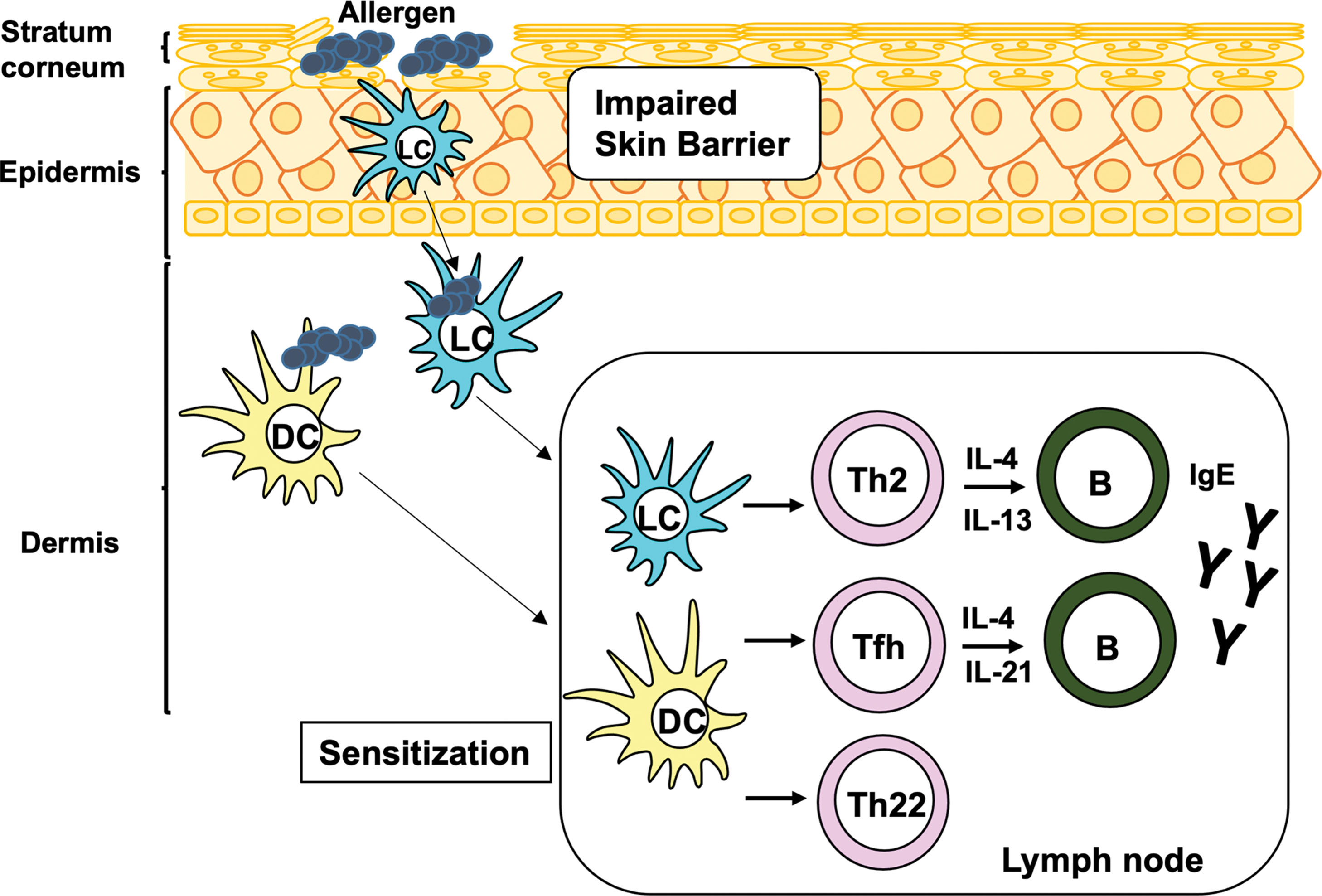
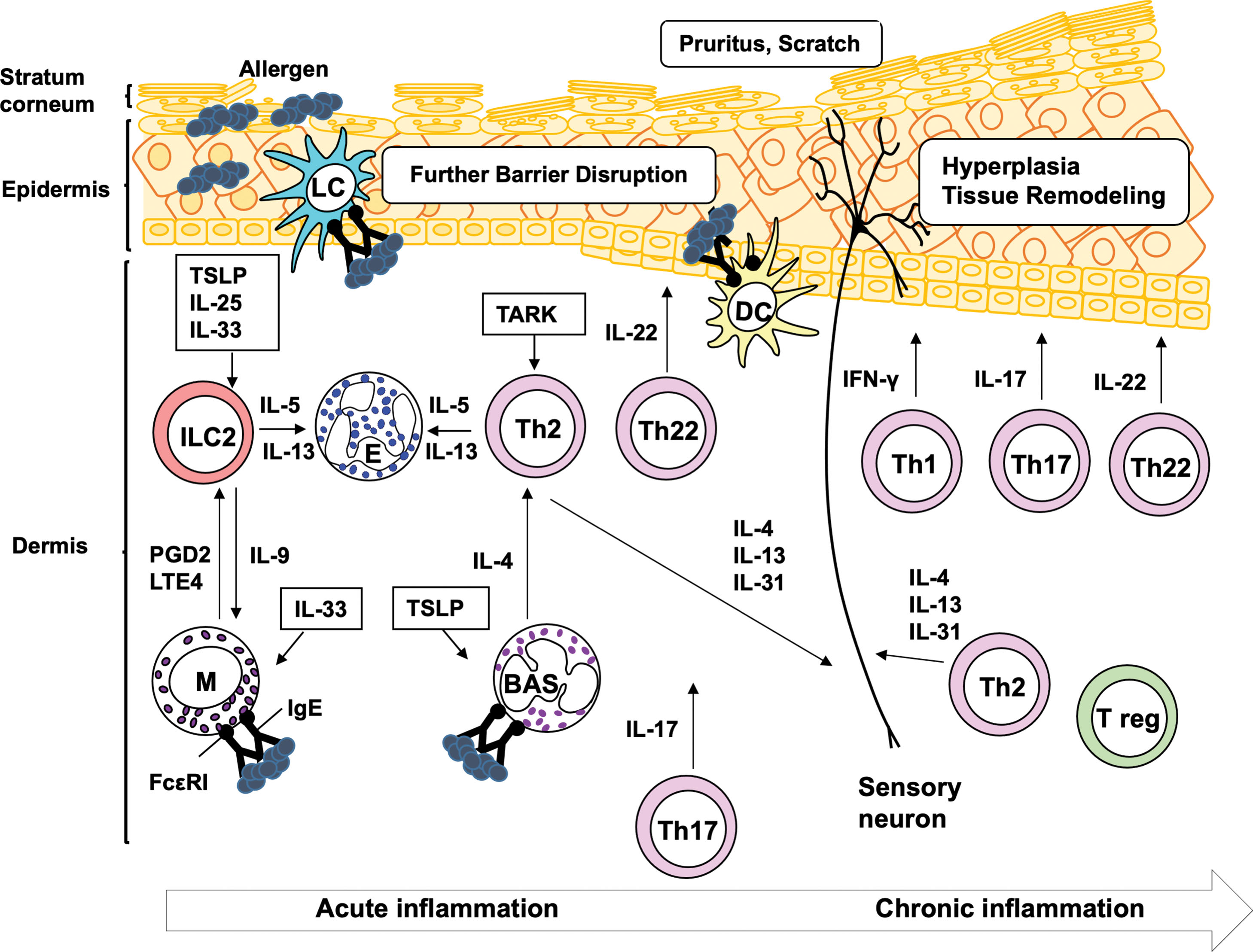
Atopic dermatitis (AD), also known as atopic eczema, is a chronic inflammatory skin disease characterized by impaired skin barrier function, marked inflammatory infiltration of activated leukocytes, intense itching, and eczematous lesions. AD is a heterogeneous disease with several subtypes, involving multiple CD4 + helper T-cell (Th) subsets and inflammatory cells. The pathologic mechanism of AD is divided into three phases: sensitization, acute inflammation, and chronic inflammation. During sensitization, specific T cells and immunoglobulin E (IgE) antibodies are produced in response to allergen(s). The first step of sensitization is allergen capture by antigen presenting cells (APCs), such as epidermal Langerhans cells (LCs) and dermal dendritic cells (DCs), which are mainly located in epidermis and dermis, respectively. This induces the activation of Th2, Th22 cells, and follicular Th (Tfh) cells, which are subsets of CD4 + Th cells ( Fig. 14.1 ). The cytokines produced by Th2 and Tfh cells, as a result of T cell–B cell interaction, trigger IgE production in B cells and elicit acute inflammation in adult AD patients ( Fig. 14.2 ). Evidence has suggested that Th2 cells also play a role in itching sensation and scratching behavior. Th22 cells cause diffuse epidermal hyperplasia. In acute inflammation, a complex interplay among several inflammatory cells, such as mast cells, granulocytes (basophils and eosinophils), and group 2 innate lymphoid cells (ILC2), is induced by both IgE-associated and -nonassociated mechanisms (see Fig. 14.2 ). In chronic inflammation, multiple subsets of CD4 + Th cells, including Th1, Th2, Th17, and/or Th22 cells, together with inflammatory cells, are accumulated in the lesional tissues (see Fig. 14.2 ). Th1 and Th17 cells aggravate inflammation and tissue remodeling, whereas Th22 cells deteriorate diffuse epidermal hyperplasia. Notably, recent studies have suggested that Th17 cells are also involved in the onset of acute inflammation in pediatric patients, indicating that pathology of AD is complex ( ). In this chapter we will review how these T-cell subsets, APCs, and inflammatory cells contribute to the development of AD. Humoral factors are reviewed in Chapter 13 .
Antigen presenting cells
APCs are a group of immune cells that are capable of processing and presenting antigens for recognition by T cells to initiate the adaptive cellular immune responses. Professional APCs in human skin include DCs, LCs, and B cells. Dermal DCs and LCs are central to the skin APC networks by physically interacting with neighboring cells, including keratinocytes, mast cells, and sensory nerve fibers for antigen acquisition and transfer, as well as signal activation and delivery, which collectively translate into skin T-cell responses. In AD, those APCs play a key role in the induction of pathogenic inflammatory T cells.
Dendritic cells
DCs are derived from hematopoietic progenitor cells in the bone marrow. They express various types of receptors for pathogen-associated pattern recognition molecules (PAMPs) for dictating pathogenic insults. Once they are activated by PAMPs, DCs upregulate major histocompatibility complex (MHC) molecules and costimulatory receptors, and act as the major professional APCs in establishing immunity against pathogens. Alternatively, upon sensing certain ligands such as those binding to C-type lectins carrying an immunoreceptor tyrosine-based inhibitory motif (ITIM) (e.g., DC-SIGN), they mature into suppressive DCs actively inducing tolerance against self-components, commensal microbacteria, and environmental antigens ( ). Recent high-dimensional phenotypic mapping of human DCs revealed that human dermal DC subsets are distinct from blood and lymphoid tissues, exhibiting interindividual variation ( ). Dermal DCs are grouped into multiple phenotypic and distinct functions, which include conventional DC1s (cDC1s), cDC2s, and monocyte-derived DCs ( ). Based on the genetic ablation approach in mouse models, primary functions of DC subsets have been defined. cDC1s (CD141 + DCs) are essential for cross-priming and Th1 responses, whereas cDC2s (CD1c + DCs) promote Th2 immune responses ( ). Under inflammatory conditions, monocyte-derived DCs are differentiated in the epidermis and act as local APCs along with LCs (see next section) ( ). Dermal DCs not only locally can activate tissue resident T cells within the skin but also carry acquired antigens to skin-draining lymph nodes where they prime naïve and memory T cells.
In AD, DCs mature in the presence of a cytokine, thymic stromal lymphopoietin (TSLP), which plays a prominent role in the induction of inflammatory Th2 responses. TSLP is highly expressed by keratinocytes in AD lesions, which is also associated with LCs activation and migration ( ). In vitro, TSLP strongly up-regulates the expression of MHC class II and costimulatory molecules, including OX40L on DCs ( ). TSLP-matured DCs produce CC chemokine ligand 17 (CCL17; also known as thymus and activation-regulated chemokine [TARC]) and CCL22, which recruit pathogenic Th2 cells in AD but fail to produce Th1 polarizing cytokine interleukin-12 (IL12) or proinflammatory cytokines ( ). Furthermore, TSLP-matured DCs can induce robust proliferation of naïve CD4 + T cells, which subsequently differentiated into Th2 cells that produce allergy-associated cytokines IL4, IL5, IL13, and tumor necrosis factor (TNF), but not IL10 and interferon-gamma (IFN-γ) ( ).
Langerhans cells
LCs are highly unique professional APCs that reside in the epidermis under steady conditions, and make up 3% to 5% of all nucleated cells in the epidermis ( ). LCs originate from tissue-resident macrophage precursor cells that directly seeded to the epidermis during embryonic development ( ) and are maintained by self-renewal mechanism via autocrine transforming growth factor-β (TGF-β) production ( ). Despite their macrophage origin as defined by a lineage-specific transcription factor Mafb expression, LCs share many phenotypic features with cDCs. For example, they have migratory capacity to draining lymph nodes via CC chemokine receptor (CCR) 7–mediated signals and express MHC class II as well as CD1a and CD1c that are MHC class I–related molecules involved in lipid antigen presentation ( ). A recent lineage tracking study indeed shows that LCs express Zbtb46, the transcription factor enforcing cDC identity ( ).
Under homeostatic conditions, LCs are thought to constantly migrate to the lymph nodes and present self antigen to establish immune tolerance ( ). Also, as a part of the first line of defense to pathogens, they protrude their dendrites via tight junctions toward the stratum corneum and can acquire antigens across epidermis ( ). After barrier disruption, such as in the case of AD or in patients with filaggrin deficiency, LCs exhibit activated phenotype with increased proliferation and expression of costimulatory molecules: CD80, CD86 ( ), and FcεRI ( ), the high-affinity IgE receptor that binds allergen-specific IgE thereby promoting allergen deposition and uptake ( ). In subsequent bacterial infection in the AD lesions, LCs likely mediate Th17 responses to certain bacteria. For example, in a mouse model of AD, infiltrating γδT cells and Th17 cells in response to Staphylococcus aureus were abrogated when mice lacked LCs ( ). Human LCs have also been shown to induce IL22 production by γδT cells, clearing S. aureus infection ( ).
B cells
As discussed in Chapter 13 , B cells act as APCs to support differentiation of Tfh cells, leading to high-affinity allergen-specific IgE production. In addition, B cells can modulate immune response through cytokine productions. Evidence suggests the existence of skin-specific B-cell subsets with restricted usage of heavy chain V gene and lower representation of IgG1 compared to bloodborne counterparts. These B cells may play a role in skin homeostasis by regulating wound healing and cutaneous microbiome through production of various cytokines such as IL6, granulocyte macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL4, and IL10 ( ). Evidence has suggested the existence of skin-specific B-cell subsets that may play a role in skin homeostasis by regulating wound healing and cutaneous microbiome through production of cytokines such as IL6, IL10, TGF-β, platelet-derived growth factor, and basic fibroblast growth factor ( ). In addition, skin-associated B1-like regulatory B cells have emerged as critical negative regulators of skin inflammation via IL10 production.
T cells
T cells are a subset of lymphocytes generated in the thymus, playing a central role in the adaptive immune system. They express T-cell receptor (TCR) with diverse specificities and recognize antigenic peptides bound to MHC presented by antigen presenting cells. In the thymus, the self-tolerance mechanism ensures elimination of high-affinity autoreactive T cells that recognize self antigens ( ). T cells (CD4 + and CD8 + T cells) that survived through the selection enter the peripheral T-cell pool or reside in peripheral lymph nodes and tissues, including the skin. There, naïve CD4 + T cells differentiate into functionally distinct and diverse effector T cells (such as Th1, Th2, Th17, and Tfh cells), in response to polarizing immunologic milieus set by a combination of specific TCR signaling strength, and costimulatory and cytokine signals provided by APCs.
AD is considered a T-cell–driven disease carrying a complex immune signature with phenotypic variations depending on the stage, age, and race ( ). Once sensitized, the T-cell–mediated immune reaction is progressive. The acute phase, especially in childhood, is predominantly driven by an inflammatory Th2 phenotype with production of Th2 (IL4, IL5, IL13, IL31, and CCL18) and Th22 (IL22 and S100A proteins) cytokines ( ). Infiltration of activated CD45RO + (a memory marker) cutaneous lymphocyte-associated antigen (CLA + ) T cells around the perivascular area of postcapillary venules in the upper dermis is a hallmark of acute eczematous lesions ( ). This inflammatory Th2 type response is later shifted toward Th1 and Th17 dominant responses in the chronic stage with increased chemokine (C-X-C motif) ligand 10 (CXCL10) and IFN-γ production ( ). In addition, Tfh cells that help to produce IgE and regulatory T cells (Treg) to suppress T-cell responses are also likely involved in AD.
Inflammatory Th2 and Th22 cells
AD has been classically thought to be a Th2 dominant disease. Th2 cells that accumulate in the upper dermis and later penetrate the basal membrane into the epidermal keratinocytes layer are mainly mediated by Th2 cytokines such as IL4, IL5, IL13, IL31, and IL33. IL4 and IL13 induce IgE production by B cells, whereas IL4, IL5, IL13, and IL33 are maturation, activation, and/or migration factors for mast cells, basophils, and eosinophils. These Th2 cytokines as well as IL22 from Th22 cells have been shown to influence keratinocyte differentiation by inducing keratinocyte apoptosis and reducing filaggrin and tight junction proteins (such as claudins), which collectively results in skin barrier defect ( ). These Th2 and Th22 cytokines also inhibit skin production of antimicrobial peptides ( ) and initial development of Th1 and Th17 response effective for preventing infections ( ), which accelerates the epidermal barrier disruption and enhances the risk of S. aureus infection in AD skin.
Th22 cells are identified as IL22 + IFN-γ − IL4 − and IL17 − CD4 + T-cell subset with expression of CCR6 as well as skin homing receptors CCR4 and CCR10 ( ). Significant increase in IL22 levels is reported in acute legion of AD ( ). In addition, the numbers of Th22 (CD4 + IL22 + IL17A − IFN-γ − ) cells as well as Tc22 cells (CD8 + IL22 + IL17A − IFN-γ − ) in the chronic lesional skin are significantly increased compared to those seen in psoriasis disease ( ). A recent study has shown that Th22 cells inhibit staphylococcal enterotoxins, whereas Tc22 cells exhibit an enhanced response to the bacterial stimuli, indicating the relevance of CD8 + T cells modulated by staphylococcal enterotoxins in adult AD patients causing the immunologic imbalance ( ).
How biased Th2 differentiation is induced in the first place remains unclear. It is likely multiple genetic, cellular, and cytokine/chemokine factors, including TSLP, are involved in conditioning Th2 differentiation ( ). A recent study shed light on the role of ionic checkpoints, such as sodium chloride (NaCl) and potassium, as cutaneous microenvironmental checkpoint factors that may influence the Th2 polarization in AD ( ).
Based on cytokine profile, the Th2 response seen in patients with AD is an “inflammatory” Th2 type, different from conventional Th2 response ( ). The initial acute inflammatory Th2 response in AD is marked by high levels of TNF and conventional Th2 cytokines (IL4, IL5, and IL13) but little or no IL10 ( ). The inflammatory Th2 response is driven by DCs and LCs matured in the presence of TSLP that is highly expressed by keratinocytes, mast cells, epithelial cells, and fibroblasts in AD lesions. In addition, TLSP-stimulated DC and LC promote production of CCL17 and CCL22 that recruit pathogenic Th2 cells. CCL17 and CCL22 are also expressed by dermal endothelial cells ( ). CCR4 and its ligands are regarded as potential therapeutic targets for AD (see Chapter 13 ).
Finally, Th2 cells play a key role in AD-related pruritus through IL4, IL13, and IL33 production. These Th2 cytokines directly activated both human and mouse sensory neurons to transmit itch signal from the peripheral to central nervous system ( ). Transgenic mouse models overexpressing IL4 and IL13 exhibited pruritic dermatitis along with symptoms associated with AD such as thickening lesions and increase in IgE ( ). Neurosensory mechanisms in AD are further discussed in Chapter 16 .
Th1 and Th17 cells
In the later chronic stage of adult AD patients, T-cell response is shifted toward Th1 and Th17 response. Th1 cells, marked by the expression of T-bet, are polarized from naïve T cells in the presence of DCs secreting large amounts of IL12p70. Th1 cells that produce IFN-γ are seen infiltrating in chronic lesions colonized by S. aureus and cause skin tissue damages. Skin-colonized S. aureus produces lipoteichoic acid, a toll-like receptor 2 (TLR2) agonist ( ). DCs matured with lipoteichoic acid plus IL4 in vitro produce IL12p70 and IL23 to promote Th1 and Th17 priming, respectively ( ). Th17 cells are characterized to produce IL17 and IL22 ( ). In AD, the numbers of IL17- producing T cells in peripheral blood and lesions correlates with disease severity ( ). IL17 production by Th17 cells requires staphylococcal superantigen stimulation, indicating the role of bacterial exposure in triggering a Th17 response in AD ( ). Collectively, these studies suggest that IL12, which is often produced by microbial infections, plays a critical role in the shift toward Th1/Th17 response by overriding the OX40L-induced inflammatory Th2 response driven by TSLP ( ). Notably, showed that accumulation of Th17 cells was observed in AD patients aged 3 months to 5 years with early onset in the previous 6 months and moderate-to-severe disease. The same research group showed the significant Th17/Th22 skewing in pediatric AD with early onset but lacked the Th1 upregulation that characterizes adult AD ( ). Taken together, the pathologic mechanism for onset and development of AD appears different in pediatric and adult patients and has to be elucidated in more detail.
T follicular helper cells
Tfh cells marked by high levels of CXCR5, PD-1, and Bcl-6 expression are specialized to support B-cell response in germinal centers that are essential for class switching recombination and somatic hypermutation for the generation of high-affinity antibody producing plasma cells and memory B cells ( ). By supporting humoral response, Tfh cells are involved in clearing infections but are adversely linked to autoimmunity. In AD, Tfh cells are thought to support allergen-specific IgE production. Notably, recent studies have suggested that IgG1 and IgE response is mainly supported by IL4 produced by Tfh cells but not by Th2 cells ( ). In addition, a recent study showed that differentiation of Tfh cells are promoted by TSLP-matured DCs via OX40L, but not ICOS, costimulation. Such TSLP-DC differentiated Tfh cells are capable of helping IgE secretion by memory B cells through production of IL4, IL21, and CXCL13 ( ). Evidently blood-circulating Tfh cells are increased in AD compared to healthy children, but not adults, implying that the role of Tfh cells in children and adults is different ( ).
Regulatory T cells
Tregs are identified as CD4 + CD25 hi Foxp3 + cells that establish and maintain immune homeostasis by suppressing immune responses ( ). Tregs are generated during thymic T-cell development as a subset of self-reactive T cells under transcription factor Foxp3 regulation. Tregs are specialized in regulating inflammatory responses by utilizing tissue-dependent distinct mechanisms such as IL10 secretion and TGF-β–mediated contact dependent suppression. In skin, Foxp3 + Tregs are one of the largest immune cell subsets; they localize to hair follicles where commensal bacteria reside ( ). Accordingly, the frequency of Tregs fluctuates 20% to 80% of total CD4 + T cells at steady state along with the hair follicle cycle ( ). Skin-resident Tregs express skin-homing receptors CLA (Psgl-1), CCR4, and CCR6 as do pathogenic Th2 cells in AD. The ligands for CCR4 (i.e., CCL17 and CCL22) are expressed by dermal endothelial cells ( ). Although the role of Tregs in AD disease is not entirely clear, it is conceivable that they play a critical role in regulating abnormal inflammatory Th2 response based on the spontaneous development of AD-like eczematous dermatitis in patients with the IPEX syndrome that have dysfunctional Tregs due to the mutations in the Foxp3 gene ( ). In a mouse model of AD, retinoic acid receptor–related orphan receptor-α (ROR-α)–expressing Tregs ( ) and IRF4-expressing Tregs ( ) in skin were shown to be important in regulating type 2 immune responses. Finally, dysregulation of Tregs functions has been suggested in AD. The frequency of Tregs is elevated in both mouse models and human patients and correlates with AD severity, indicating that Tregs in AD are unable to appropriately downregulate the ongoing inflammatory response ( ).
Autoreactive T cells
Recently, the pathologic role of autoreactive T cells in AD has been suggested ( ). Although at low frequencies (1–10 in 10 6 T cells), harmful autoreactive T cells that escaped central tolerance in thymus are detectable in the peripheral T-cell pool ( ). Their response is normally suppressed via multiple mechanisms, including anergy, ignorance, and suppression by Tregs ( ). However, a combination of genetic and environmental factors may trigger their inappropriate activation, resulting in autoimmune responses ( ). In fact, autoreactivity has been known in inflammatory skin diseases, including AD, but the role of autoreactive T cells differs among diseases ( ). In the case of vitiligo, autoreactive T cells are directly involved as effector T cells by attacking the target in skin or as memory cells (i.e., tissue resident memory T cells [T RM ]) that are thought to be responsible for relapsing disease. In pemphigus and pemphigoid, autoreactive T cells help B cells to produce pathogenic autoantibodies to cause skin damage. Autoreactive T cells in AD have both functions. The presence of autoreactive IgE antibodies is seen in about 30% of the AD patients ( ). Several IgE-binding keratinocyte-associated antigens show homology with environmental antigens, indicating that molecular mimicry may be a mechanism of autoantibody production in skin diseases ( ). Also, IFN-γ and IL4 secreting effector CD8 + T cells specific to Hom s 2, the α-chain of the nascent polypeptide-associated complex (α-NAC), are found at higher numbers in AD patients compared to non-AD controls ( ).
Mast cells
Mast cells are long-lived, tissue-resident cells with one nucleolus and many granules. Mast cells express tetrameric αβγ2 form of the high-affinity IgE receptor FcϵRI on the cell surface. Degranulated mast cells are observed in lesional skin of patients with acute AD. Moreover, the number of the degranulated cells is often increased in chronic AD, suggesting that IgE-mediated pathology may be a driving force of AD ( ). The association of allergen with IgE bound to FcϵRI triggers degranulation and activation of mast cell degranulation and activation. Mast cell degranulation is characterized by the release of bioactive chemical mediators (e.g., histamine, serotonin, substance P, and heparin) and mast cell–specific proteases (e.g., tryptases, chymases, and matrix metalloproteases, in particular 1 and 4) ( ). In addition, activated mast cells release de novo synthesized proinflammatory lipid mediators (e.g., prostaglandin D2, leukotriene C4, leukotriene B4, and platelet-activating factor). These mast cell–derived components cause itching, redness, tissue damage, and skin dryness in AD patients ( ). Given the fact that histamine levels are also highly elevated in inflamed skin, it is likely that histamine plays a relevant role in disease pathology. However, antagonists that block histamine H 1 or H 2 receptors are largely ineffective in reducing chronic symptoms in AD. Recent clinical studies have reported that H 4 receptor antagonists display antipruritic and antiinflammatory effects and might be a novel therapeutic option in AD treatment ( ).
FcεRI-engaged mast cells also produce a large variety of (i) cytokines, including interleukins (IL1β, IL3, IL4, IL5, IL6, IL8, IL9, IL10, IL11, IL12, IL13, IL15, IL16, IL18, IL22, IL25, and IL33), TNF-α, TGF-β, and GM-CSF; (ii) growth factors, including fibroblast growth factor 2, vascular endothelial growth factor, and nerve growth factor; and (iii) CC and CXC chemokines, including CCL2 (monocyte chemotactic protein 1) and CCL3 (macrophage inflammatory protein-1α) ( ). Mast cell–derived cytokines and chemokines cause activation and trafficking of inflammatory cells such as eosinophils and Th2 cells into the AD sites.
In addition to FcϵRI, mast cells express various types of receptors, including pattern recognition receptors (e.g., TLRs), complement receptors, cytokine receptors, and immunoglobulin receptors, all of which are involved in the activation of mast cells ( ). In the inflamed skin of patients with chronic AD, S. aureus superficially colonizes and activates mast cells via TLR2 and TLR4 ( ). IL33, an alarmin cytokine released upon epithelial barrier disruption (e.g., by allergens with protease activity, granule components released by mast cells and granulocytes, and other stimuli in allergic condition), activates mast cells via ST2 receptor ( ). Such TLR or ST2 receptor engagement augments FcϵRI-mediated mast cell degranulation, induces cytokine/chemokine production even in the absence of IgE stimulation, and promotes inflammation.
Basophils
Basophils principally circulate in peripheral blood and represent less than 1% of circulating leukocytes under homeostatic conditions. Like mast cells, basophils have many granules and express FcεRI on their cell surface. Their granules contain chemical mediators (e.g., histamine, chondroitin sulfate, and substance P) and other basophil-specific proteins (e.g., basogranulin). FcεRI engagement triggers degranulation, de novo synthesis of lipid mediators (e.g., leukotriene C4 and platelet-activating factor), and production of cytokines and chemokines in the cells ( ). In comparison to mast cells, basophils produce a limited variety of lipid mediators, cytokines, and chemokines ( ). Although basophils have recently been implicated in the pathogenesis of murine AD-like disease, their precise role in human disease remains to be further elucidated.
Basophils produce high amounts of IL4 and are considered initiators and accessory cells for the Th2 cell polarization in AD ( ). Recent studies have shown that basophil-derived IL4 promotes IL5 and IL13 secretion by ILC2, leading to eosinophilia in a murine model of AD ( ). (Detailed discussion of ILC2 is in the last section of this chapter.) Increased levels of basophils, ILC2, and/or Th2 cells in skin of AD patients has also been observed ( ). In addition, basophils proliferate and migrate into tissues to promote type 2 immune responses upon stimulation with TSLP, a key cytokine released by keratinocytes involved in the pathogenesis of AD ( ). However, basophil-deficient mice showed only modest attenuation in the development of AD-like inflammation ( ); this observation might be attributed to a limited variety of cytokine/chemokine production and a short lifespan of the cells. Mast cells have a long lifespan of 20 to 30 days, whereas basophils have a lifespan of only 2 to 3 days. Moreover, IgE is a survival factor for mast cells but not for basophils ( ). Therefore the contribution of IgE- and/or TSLP-activated basophils to the pathogenesis of AD still needs to be elucidated.
Eosinophils
Eosinophils represent 1% to 6% of circulating leukocytes in blood and do not migrate to the skin under normal physiologic conditions. However, increased blood and tissue eosinophilia are often associated with disease severity of AD ( ). Eosinophil degranulation and deposits of granular proteins in lesional skin of AD patients have been observed ( ). Secretory granules of eosinophils contain mainly five cytotoxic proteins, including the eosinophil major basic protein, eosinophil-cationic protein (ECP), eosinophil protein X (EPX), eosinophil-derived neurotoxin (EDN), and eosinophil peroxidase (EPO). These cytotoxic proteins are implicated in cytotoxicity, tissue damage, and mediation of AD since several studies showed that their levels in sera and urine samples correlate with disease progression in AD patients ( ).
Cytokines and chemokines that are involved in the development of skin eosinophilia include IL3, IL5, IL13, IL33, GM-CSF, CCL11 (eotaxin-1), CCL24 (eotaxin-2), and CCL26 (eotaxin-3) ( ). Increased expression of IL5, IL13, IL33, CCL11, CCL24, and/or CCL26 in lesional skin of AD patients has been reported. Among the chemoattractants, IL5 exhibits the most potent eosinophil-specific properties to induce maturation, migration, activation (that leads to release of granules), proliferation, and survival of the cells. Several studies have shown the association of IL5 expression levels with eosinophilia in lesional skin ( ). However, two single doses of 750 mg mepolizumab, an antibody against IL5, did not meet clinical expectations in a short-term pilot study in patients with modest to severe AD, although a significant decrease of blood eosinophils was reached ( ). These data trigger the debate whether eosinophils could serve as therapeutic targets for treatment of AD. To address the long-term risk:benefit ratio of mepolizumab, new studies have been undertaken in a subset of patients with modest to severe AD and increased eosinophil counts ( ).
ILC2
ILCs are the most recently identified immune lymphoid cells that are phenotypically similar to Th subsets but lacking T- or B-cell receptors ( ). There are three main subsets of ILCs: ILC1, ILC2, and ILC3, which produce IFN-γ, IL5/IL9/IL13, and IL17A/IL22, respectively. These ILC subsets are considered to mirror the profile of cytokine production of Th1, Th2, and Th17 cells. Although ILC population is rare, the cells are capable of producing significantly higher amounts of cytokines than T cells. Evidence has suggested that ILC2 plays a crucial role in the development of AD by producing IL5, IL9, and IL13. The levels of ILC2 are increased in lesional skin, but not in peripheral blood, of adult AD patients, when compared to healthy subjects and patients with psoriasis ( ).
ILC2 expands and produces cytokines upon stimulation with IL25, IL33, and TSLP individually, or in combination ( ). These cytokines are alarmin cytokines released by damaged epithelial cells and keratinocytes in AD condition. Increased numbers of ILC2 were associated with expression levels of IL25, IL33, and TSLP in the AD tissues ( ). IL25- and IL33-induced cytokine production of ILC2 is augmented by prostaglandin D2 and leukotriene E4 derived from mast cells. IL4 derived from basophils also promotes the cytokine production of ILC2 ( ).
The depletion of murine ILCs significantly ameliorates skin inflammation in a model of AD-like inflammation, suggesting that ILC2 plays an essential role in promoting skin inflammation ( ). The potential of ILC2 to produce IL5 and IL13 implies a role in induction of eosinophilia, barrier dysfunction, and causing an itching sensation in AD. ILC2 is also a key source of IL9, a mast cell growth factor, suggesting that these cells may affect disease severity by regulating mast cell number and/or function in the skin. Live imaging studies showed a proximity of dermal ILC2 with skin-resident mast cells in mice ( ). However, the majority of studies investigating the functions of this cell population have been based on murine models. Detailed characterization of ILC2 in lesional tissues of AD patients is essential to deepen our insights into pathologic mechanisms of AD and to establish a novel strategy for AD treatment.
Summary
AD was initially recognized as a Th2-driven skin inflammatory disease. However, accumulating studies have revealed that AD is a more complex and heterogeneous disease with several subtypes, involving multiple CD4 + T-cell subsets (e.g., Th1, Th2, Th17, and Th22), APCs (e.g., DCs, LCs, and B cells), innate inflammatory cells, keratinocytes, and possibly neuronal cells for pruritus. Several classes of newly identified cells exist, of which roles need to be clarified. For example, the relevance of Tfh cells in AD needs to be addressed in relationship to IgE production. The presence of autoreactive T cells in AD patients raises a possibility that AD is associated with autoimmune diseases. ILCs, which have been identified in mice, need to be characterized in AD patients. It is also crucial to identify T-cell phenotypes in pediatric AD since they appear to be different from those in adult patients. Current advances in technologies, such as the single cell sequencing approach, may further allow characterization of key cellular components in AD patients and relevant mouse models, which would improve the understanding of the pathophysiology of AD. In the meantime, a large-scale transcriptome analysis and machine-learning data mining may help identify essential signaling pathways shared among multiple cell types in AD as therapeutic targets.
Further readings
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


