Abstract
This chapter discusses benign epidermal tumors, ranging from extremely common lesions (e.g. seborrheic keratoses) to less common (e.g. disseminated superficial actinic porokeratosis) to unusual or rare (e.g. warty dyskeratoma, clear cell papulosis). Seborrheic keratoses are seen in the vast majority of older adults; the lesions are papular or verrucous, appear “stuck on”, and vary in color from white to tan–brown to black. A number of entities are related to seborrheic keratoses, including solar lentigines, stucco keratoses, and dermatosis papulosa nigra. The multiple types of porokeratosis are reviewed as are examples of nevi that follow the lines of Blaschko (e.g. epidermal nevus, inflammatory linear verrucous epidermal nevus, nevus comedonicus). Acanthosis nigricans and confluent and reticulated papillomatosis, examples of epidermal proliferation, are also described.
Keywords
solar lentigo, solar lentigines, seborrheic keratosis, seborrheic keratoses, lichenoid keratosis, dermatosis papulosa nigra, stucco keratoses, porokeratosis, Flegel disease, acrokeratosis verruciformis, cutaneous horn, clear cell acanthoma, inverted follicular keratosis, warty dyskeratoma, large cell acanthoma, epidermal nevus, inflammatory linear verrucous epidermal nevus (ILVEN), nevus comedonicus, acanthosis nigricans, confluent and reticulated papillomatosis, clear cell papulosis
The majority of entities in this chapter can be further classified according to the algorithm in Fig. 109.1 .
Solar Lentigo
▪ Lentigo senilis ▪ Liver spot ▪ Senile freckle
- ▪
Tan to brown to black macules in sites of exposure to UVR
- ▪
Histologically, the rete ridges often have club-shaped or bud-like extensions, in addition to increased basal layer pigmentation
- ▪
Solar lentigines and macular seborrheic keratoses exist along a continuum
Epidemiology
Solar lentigines are present in 90% of Caucasians over the age of 60 years, and their incidence increases with advancing age. They also occur, but less often, in younger individuals and in Asians as a consequence of acute or chronic sun exposure. Inherited patterned lentiginosis favors more lightly pigmented African-Americans, including those with mixed Native American (Indian) heritage.
Pathogenesis
Solar lentigines result from epidermal hyperplasia, with variable proliferation of melanocytes and accumulation of melanin within keratinocytes, in response to chronic exposure to UVR. Similar to seborrheic keratoses, activating somatic mutations in FGFR3 and PIK3CA have been detected in solar lentigines ( Table 109.1 ) . As with ephelides, variants in the melanocortin-1 receptor gene are also linked to the development of solar lentigines .
| SOMATIC ACTIVATING MUTATIONS IN BENIGN EPIDERMAL TUMORS | |
|---|---|
| Tumor | Gene(s) |
| Solar lentigines | FGFR3 * , PIK3CA ** |
| PUVA lentigines | BRAF |
| Seborrheic keratosis | FGFR3 , PIK3CA |
| Lichenoid keratosis † | FGFR3 , PIK3CA , HRAS > KRAS |
| Dermatosis papulosa nigra | FGFR3 |
| Stucco keratosis | PIK3CA |
| Epidermal nevi with hyperkeratosis, acanthosis and papillomatosis (but no epidermolytic hyperkeratosis) histologically; also referred to as “common” or verrucous epidermal nevi or keratinocytic nevi | FGFR3 , PIK3CA , HRAS > NRAS > KRAS |
| Acneiform nevus with hypopigmented background skin (Munro acne nevus) | FGFR2 |
| Nevus comedonicus | NEK9 |
| See Table 62.7 for genes underlying nevus sebaceus and epidermal nevi with epidermolytic hyperkeratosis, acantholytic dyskeratosis, or associated overgrowth syndromes | |
* Germline mutations in this gene lead to skeletal dysplasia syndromes associated with acanthosis nigricans.
** Germline mutations in this gene are often lethal, but occasionally can lead to a Cowden syndrome-like phenotype.
† Related to benign lentigo, SK or actinic keratosis; the latter can have RAS mutations.
Clinical Features
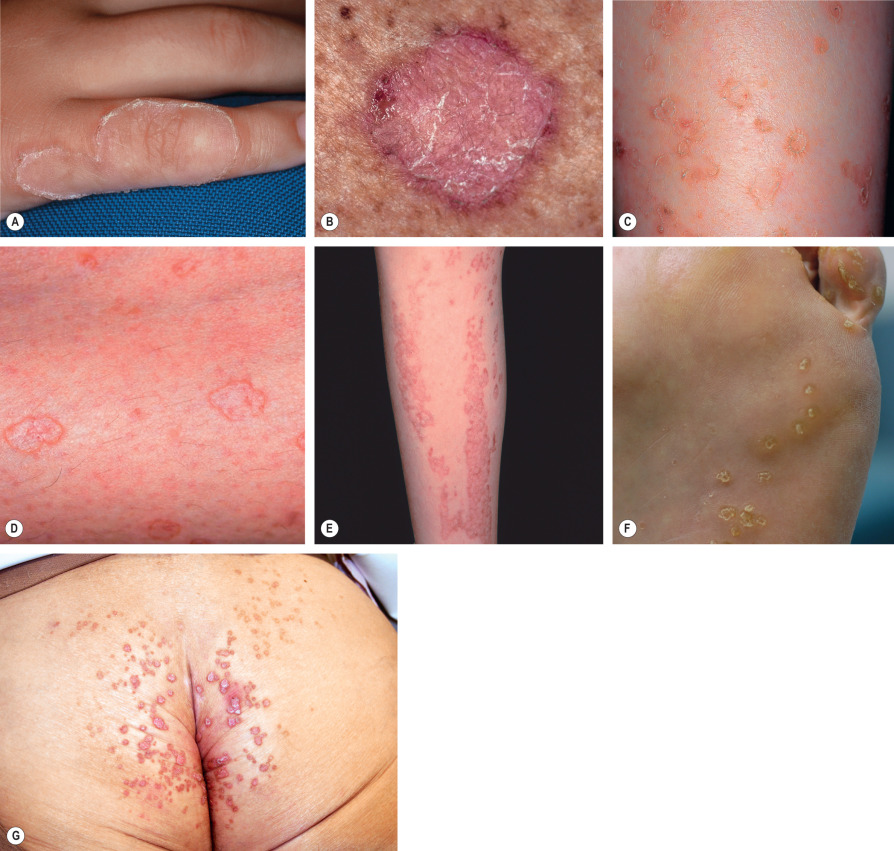
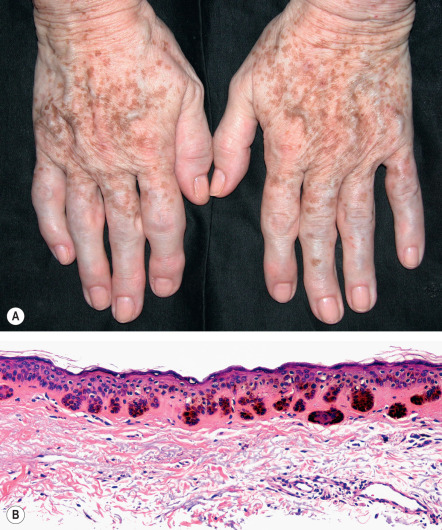
Solar lentigines are well-circumscribed, round, oval or irregularly shaped macules that vary in color from tan to dark brown or black. More lightly pigmented lesions are usually homogeneous, whereas darker ones tend to have a mottled appearance. Solar lentigines almost always present as multiple lesions. They usually vary in diameter from 3 mm to 2 cm and can become confluent. Solar lentigines occur in sun-exposed sites, predominantly the dorsal aspects of the hands and forearms, face, upper trunk, and shins ( Fig. 109.2A ).
By tangential lighting, a reticulated pigment pattern may be appreciated, and Wood’s lamp illumination enhances both visible and inapparent lesions. The dermoscopic features of a solar lentigo may include the following: a diffuse light brown structureless area, sharply demarcated and/or moth-eaten borders, fingerprinting, and a reticular pattern with thin lines that are occasionally short and interrupted . A particular variant that has been termed hypermelanotic or “ink spot” solar lentigo is characterized by a striking jet-black color and a stellate outline. By dermoscopy, a black branching pattern is seen.
While solar lentigines may fade slightly after cessation of exposure to UVR, they usually persist indefinitely. Solar lentigines have been shown to be an independent risk factor for the development of cutaneous melanoma. Histologic overlap suggests that both lichenoid keratoses and reticulated seborrheic keratoses can evolve from solar lentigines.
Associated Diseases and Special Forms
A PUVA (psoralen + UVA) lentigo is a well-defined hyperpigmented macule that commonly develops in individuals undergoing long-term PUVA photochemotherapy (see Ch. 134 ) . These lentigines can arise in any site exposed to UVA light and often persist for years after discontinuation of PUVA therapy. In contrast to solar lentigines, PUVA lentigines usually exhibit a darker brown color and more of a stellate appearance, clinical features that may suggest cutaneous melanoma. Histologically, PUVA lentigines display lentiginous hyperplasia of large melanocytes that often exhibit mild cytologic atypia , and they often contain BRAF mutations . Numerous solar lentigines are also seen in patients with xeroderma pigmentosum, and they are often an initial cutaneous finding (see Ch. 87 ).
Pathology
The rete ridges often have club-shaped or bud-like extensions ( Fig. 109.2B ), and branching or fusing of rete ridges, forming a reticulated pattern. The epidermis between the rete ridges may appear thinned. There is increased basal layer pigmentation, especially in the basaloid cells of the rete ridges, and the lesional keratinocytes are larger than those of adjacent, non-involved epidermis . In some (but not all) cases, the melanocytes are slightly increased in number. Melanocytes in DOPA-stained sections of solar lentigines exhibit increased melanogenesis, and these cells have more numerous as well as longer and thicker dendritic processes than the melanocytes of normal skin. The superficial dermis often contains melanophages and occasionally a mild perivascular infiltrate of lymphocytes. Solar elastosis is also prominent. Electron microscopic studies reveal abundant melanosome complexes in keratinocytes which appear to be larger than in normal surrounding skin.
Differential Diagnosis
The differential diagnosis of a solar lentigo includes an ephelid, macular seborrheic keratosis, pigmented actinic keratosis, lentigo maligna, simple lentigo, junctional melanocytic nevus, and large cell acanthoma. Table 112.1 outlines the similarities and differences between solar lentigines and ephelides. There is essentially a continuum extending from solar lentigo to macular seborrheic keratosis. Demonstration of a keratotic surface with horn cysts is consistent with seborrheic keratosis. Pigmented actinic keratosis is more likely to have a scaly rough surface. Lentigo maligna often exhibits greater variation in pigmentation and irregularity of borders compared with solar lentigo, and by dermoscopy, rhomboid structures and grey dots can be seen, in particular on the face. Simple lentigines are often smaller and more heavily pigmented than solar lentigines, and they arise during childhood with less relationship to UVR exposure (see Ch. 112 ).
Treatment
Although a solar lentigo is a benign lesion of cosmetic concern only, it is an indication of chronic UVR exposure which dictates monitoring of the patient for cutaneous carcinomas. Bleaching agents (e.g. hydroquinone) are not effective. Cryotherapy and laser surgery have been shown to be equally effective , but caution must be used to prevent post-treatment dyspigmentation. Preventive measures include sunscreens, physical barriers, and limitation of sun exposure .
Seborrheic Keratosis
- ▪
Common benign lesions that typically begin to appear during the fourth decade of life
- ▪
Develop anywhere except mucous membranes, palms and soles
- ▪
Tan to black, macular, papular or verrucous lesions; often have a waxy “stuck-on” appearance
- ▪
May become irritated or inflamed
- ▪
Large variation in clinical appearance and can simulate melanocytic neoplasms
Introduction
Seborrheic keratoses (SKs) are common benign skin lesions that are almost ubiquitous among older individuals. They develop in hair-bearing skin, invariably sparing the mucosal surfaces and the palms and the soles. Thus, SKs can appear on the head and neck, extremities, and trunk, especially the upper back and submammary region (in women).
Many clinical and histologic variants of SK have been described, and although they are usually easily recognized clinically, some lesions may prove difficult to diagnose by inspection alone, so biopsy for histopathologic examination may be required. This is especially true when there is a history of recent change or if there is inflammation. Larger dark lesions are sometimes biopsied when there is concern about the possibility of melanoma, but with the advent of dermoscopy, this occurs less frequently.
History
The date of the initial description of SKs is uncertain. In 1927, Freudenthal delineated their clinical and histologic features.
Epidemiology
Autosomal dominant inheritance with incomplete penetrance has been postulated to explain an apparent familial predisposition. Despite their frequent occurrence, there are few statistics on prevalence, gender or racial predilection, or geographic distribution. Almost all epidemiologic studies have noted SKs as coincidental findings. They have been reported to be more common in Caucasian populations and to affect men and women equally . Their appearance prior to the fourth decade is uncommon. Usually, lesions continue to develop throughout one’s lifetime.
Pathogenesis
Although SKs are common in areas covered by clothing, sun exposure has been implicated in their development. Supporting evidence comes from the more frequent occurrence and earlier age of onset of SKs in individuals residing in tropical climates. An Australian study found a higher prevalence of SKs within sun-exposed areas such as the head and neck in contrast to non-sun-exposed areas in the same subjects. The authors also reported a higher frequency and an earlier age of onset in their Australian study group population compared to a UK study group .
An alteration in the distribution of epidermal growth factor receptors has been observed in SKs. Based upon analysis of androgen receptor polymorphisms, a clonal origin was detected in over half of lesions evaluated, suggesting a neoplasm rather than hyperplasia . More recently, somatic activating mutations in two genes – FGFR3 , which encodes fibroblast growth factor receptor 3 , and PIK3CA , whose protein product is the alpha subunit of phosphoinositide-3-kinase – have been demonstrated within SKs (see Table 109.1 ). The enhanced activity of these two proteins leads to increased AKT activity .
In irritated SKs, apoptosis within areas of squamous differentiation has been implicated as a cause of the irritation . Although SKs are often clinically verrucous, human papillomavirus (HPV) has been detected infrequently, except in lesions in the genital region. It is likely that these latter lesions actually represent condyloma acuminatum, as the two entities may appear similar both clinically and histologically .
Clinical Features
Although occasionally solitary, SKs more commonly present as multiple, pigmented, sharply marginated lesions. They may be macules, papules or even plaques, depending on their stage of development ( Fig. 109.3 ). Even within the same lesion there may be a marked variation in color. They are usually light brown but may appear white to waxy yellow to brown–black in color. SKs typically evolve from a macule and may progress to become papular or verrucous. Keratotic plugging, a “stuck-on” appearance, and/or overlying scale are helpful features in distinguishing SKs from melanocytic neoplasms, noting that occasionally keratotic plugging can be seen in compound and intradermal melanocytic nevi.
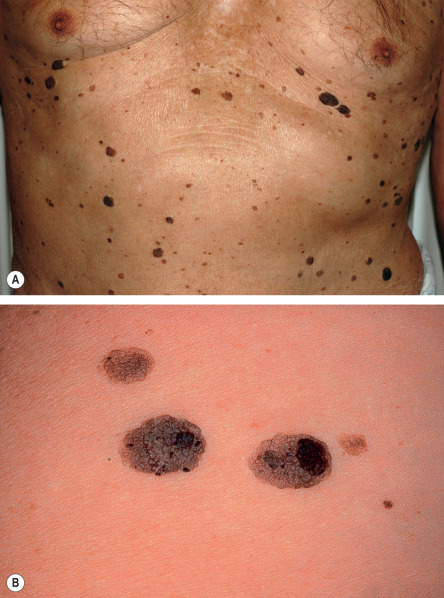
Individual lesions may be of any size but usually measure about 1 cm in diameter. They can become quite large, i.e. >5 cm in diameter. SKs may become inflamed or irritated due to friction or trauma, or rarely from secondary bacterial infection. Although usually asymptomatic, irritated or inflamed lesions may become tender, pruritic, erythematous, crusted, and rarely, pustular. Sometimes inflammation reflects an underlying diathesis, e.g. psoriasis.
Spontaneous regression, although observed, is not a common feature of SKs, even following inflammation. With trauma, the lesion may crumble, explaining reports of SKs “falling off”. Conditions associated with an abrupt appearance or increase in number of lesions, followed by regression when the condition resolves, include pregnancy , coexisting inflammatory dermatoses (in particular erythroderma) , and malignancy (next section) .
Seborrheic Keratosis and Malignancy
Instances of malignant neoplasms arising within and adjacent to SKs were reported as early as 1932 . Invasive and in situ squamous cell carcinoma (SCC), cutaneous melanoma, basal cell carcinoma (BCC), and keratoacanthoma have all been observed in association with SKs. This likely represents a coincidental neoplasm developing in adjacent skin, although it is possible that the various cell types present in an SK could develop into their respective neoplasms. In theory, the basaloid cells could give rise to BCC, the spinous cells to SCC, and melanocytes to melanoma . BCC is thought to be the most frequently associated neoplasm , although one prospective histopathologic study only identified SCC in situ in 60 (1.4%) of 4310 specimens accessioned as SK clinically . In another investigation, 4.6% of SCCs were clinically diagnosed as SKs, and in some of these, features of SK were seen in association with the SCC. The “collision” theory, when two distinct neoplasms develop separately at the same site, was entertained as a possible cause for this occurrence, especially given the prevalence of both of these lesions in the population at large . Others believe that BCCs and SKs are both derived from the infundibular portion of the hair follicle, cells of which could evolve into either neoplasm, and in some cases, both concurrently.
The sign of Leser–Trélat is a rare cutaneous marker of internal malignancy, in particular gastric or colonic adenocarcinoma, breast carcinoma, and lymphoma. It is considered to be a paraneoplastic cutaneous syndrome characterized by an abrupt and striking increase in the number and/or size of SKs occurring before, during or after an internal malignancy has been detected . Associated pruritus has been documented in over 40% of cases and the majority of the lesions are located on the back, followed by the extremities, face and abdomen . Malignant acanthosis nigricans, another paraneoplastic syndrome, may appear at the same time or shortly after the sign of Leser–Trélat in approximately 20% of patients .
Since its initial description in 1900, the validity of the sign of Leser–Trélat as a reliable marker of internal malignancy has been challenged, given the frequency of both neoplasia and SKs in the elderly population, in whom the condition is usually observed . Its description is also loosely defined, which is problematic as there are no standards for quantifying the number of lesions required for the diagnosis. The few studies investigating SKs and their link with internal malignancy have largely been inconclusive. One retrospective review examined 1752 consecutive patients with the diagnosis of SK, and of these, 62 individuals were diagnosed with an internal malignancy within 1 year before or after the diagnosis of SK. Of those 62 patients, six patients presented with findings consistent with the sign of Leser–Trélat. However, an age- and sex-matched control group demonstrated similar findings .
The pathogenesis of the sign of Leser–Trélat is uncertain, but it is thought to be related to secretion of a growth factor by the neoplasm which leads to epithelial hyperplasia , as in malignant acanthosis nigricans. To qualify as a paraneoplastic process, the cutaneous findings need to coincide with the presence of the malignancy, resolving when the primary tumor is excised or successfully treated and reappearing with its recurrence or metastasis, i.e. fulfill Curth’s postulates (see Ch. 53 ).
Pathology
There are at least six histologic types of SK: acanthotic, hyperkeratotic, reticulated, irritated, clonal, and melanoacanthoma. Different histologic features are often present in the same lesion, resulting in diverse appearances. There are varying degrees of hyperkeratosis, acanthosis and papillomatosis. Horn pseudocysts, the product of cross-sectioned epidermal invaginations, are highly characteristic, though not invariably present. Generally, the base of an SK lies on a flat horizontal plane flanked by normal epidermis. The characteristic acanthosis is the result of an accumulation of benign squamous and basaloid keratinocytes, typically projecting outward and upward in an irregular fashion. The sharp demarcation at the base of most lesions has been called the “string” sign. The marked papillomatosis and hyperkeratosis are often likened to “church spires” with shadows of retained cornified material at their peaks.
The squamous cells of many SKs are representative of those found in the normal-appearing epidermis, but some SKs may contain basaloid cells that have a smaller size, uniform appearance, and large oval-shaped nuclei. When slight intercellular edema is present, intercellular bridges are easily visualized. Cytologic atypia is usually not a feature of most SKs. Mild keratinocyte atypia and mitotic figures, when present, are usually associated with irritation and inflammation. The papillary dermis in most instances is unremarkable.
The acanthotic SK is the most common histologic type. It usually presents as a smooth-surfaced, dome-shaped papule. Slight hyperkeratosis and papillomatosis are often present, while the greatly thickened epidermis typically contains a preponderance of basaloid cells ( Fig. 109.4A ). Papillae may be narrow in some lesions, while others may be composed of interwoven aggregates of epithelial cells surrounding islands of connective tissue in a retiform pattern. Invaginated horn pseudocysts are most prevalent in this variant. Melanin is often increased in the acanthotic type of SK; it is primarily concentrated in keratinocytes and is transferred from neighboring melanocytes. Deeply pigmented lesions contain abundant melanin in basaloid cells.
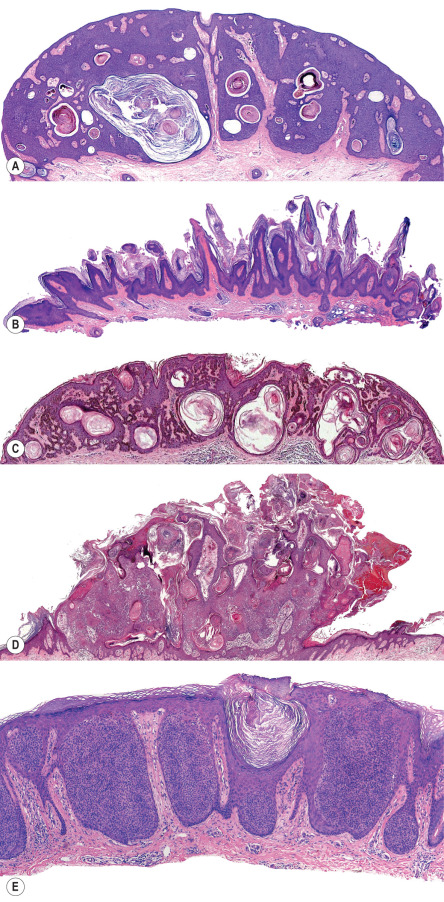
The hyperkeratotic type of SK, also known as a digitated, serrated or papillomatous type, is almost the morphologic reverse of the acanthotic type. Acanthosis is present but there is more prominent hyperkeratosis and papillomatosis ( Fig. 109.4B ). A preponderance of squamous cells relative to basaloid cells is seen. Abundant pigmentation is unusual, and keratinizing pseudocysts are observed less frequently than in acanthotic SKs. The hyperkeratotic type is the variant often described as having epidermal projections resembling “church spires”, a finding also seen in acrokeratosis verruciformis.
The reticulated or adenoid type of SK is characterized histologically by delicate strands of epithelium that extend from the epidermis in an interlacing pattern ( Fig. 109.4C ). They are composed of a double row or more of basaloid cells that may be hyperpigmented. Horn pseudocysts, although less common than in the acanthotic variant, may be observed. Often, a solar lentigo is seen at the lateral margins of the lesion, supporting the notion that reticulated SK evolves from a solar lentigo.
A lymphoid infiltrate is often present in the dermis of inflamed or irritated SKs ( Fig. 109.4D ). It may be perivascular, diffuse or lichenoid. Spongiosis is often present and there may be necrosis of keratinocytes. Squamous eddies are common findings as well. These consist of whorls of eosinophilic keratinocytes. Irritated SKs often lack the sharply demarcated horizontal base seen with most SKs.
Clonal SKs are considered by some to represent a variant of irritated SK. The clonal, or nested, type of SK is characterized by having well-defined nests of loosely packed cells within the epithelium ( Fig. 109.4E ). The nests are composed predominantly of variably sized keratinocytes that are often paler than adjacent cells and have a uniform appearance. They may also contain melanocytes.
There is a rare variant of SK with numerous basal clear cells that histologically can mimic melanoma in situ . However, these clear cells demonstrate negative immunostaining for Melan-A/MART-1 and S100 protein .
Differential Diagnosis
Most SKs are easily identified clinically, although there are entities that may have a similar appearance. Conditions thought to be variants of SKs include dermatosis papulosa nigra, stucco keratosis, and inverted follicular keratoses. They are discussed in this chapter under their respective headings. The clinical differential diagnosis includes solar lentigo, verruca vulgaris, condyloma acuminatum, Bowen disease, SCC, melanocytic nevus, melanoma, acrochordon, and tumor of the follicular infundibulum.
Clinical differentiation between macular SKs, solar lentigines, and melanocytic neoplasms such as melanoma may at times be impossible. Solar lentigines are neither keratotic nor elevated, but some are considered to represent an incipient reticulated type of SK. Over time, solar lentigines can develop into SKs as the buds of pigmented basaloid cells become thicker and there is greater acanthosis. Dermoscopy can prove useful in distinguishing these entities (see Ch. 0 ).
A review of 20 cases of verrucous melanoma demonstrated how difficult it may be to distinguish between benign and malignant tumors. Of interest, 50% of these verrucous melanomas were thought to represent SKs clinically and a histopathologic diagnosis of a benign nevus was assigned to 10%. A broad spectrum of histopathologic findings, including hyperkeratosis, pseudoepitheliomatous hyperplasia, asymmetry and exophytic papillomatous growth pattern, were present in these cases .
Acanthotic and irritated types of SK should be delineated from eccrine poroma. On occasion, SKs may demonstrate “poroid” differentiation. Clinically, poromas are skin-colored, brown, pink or sometimes red papules or nodules that may be pedunculated and multilobular. Histologically, poromas are comprised of homogeneous, small basophilic cells with a delicate fibrovascular stroma and narrow ductal lumina with eosinophilic, PAS-positive, diastase-resistant cuticles (see Ch. 111 ).
Differentiation between an irritated SK, an SCC in situ (Bowen disease), and SCC may require histopathologic examination. An irritated SK may be misdiagnosed as an SCC if atypia is present. Although squamous eddies may be abundant in both tumors, there should be no evidence of involvement of the dermis in irritated SKs. Bowen disease demonstrates a preponderance of atypical keratinocytes with vacuolated cytoplasm, close crowding of nuclei, abundant mitoses and parakeratosis. The adnexal epithelium is also involved.
The Borst–Jadassohn pattern of clonal SK is also seen in some examples of Bowen disease. In general, Bowen disease with a clonal or nested pattern has greater atypia of the individual keratinocytes.
Melanoacanthoma was originally described by Bloch and named “non-nevoid melano-epithelioma, type 1” . It is now regarded as a heavily pigmented SK by most authorities. In this variant, melanocytes are distributed throughout the lesion. While the keratinocytes contain melanin, the bulk of the pigment is present within melanocytes, many of which also have long dendrites . Although the melanocytes are prominent, there is no significant increase in their number. The heavy pigmentation of melanoacanthoma has been explained by the blockage of transfer of melanin to keratinocytes, perhaps leading to an increase in the amount of melanin within melanocytes.
Intraoral melanoacanthoma or melanoacanthosis was first described in 1979. These lesions bear a striking clinical resemblance to the melanoacanthoma type of SK. Histopathologically, they are distinct from the cutaneous variant by having only minimal epithelial hyperplasia. Their histologic features are most like those of a heavily pigmented lentigo simplex, with a proliferation of dendritic melanocytes within the basal layer of the epithelium .
Verrucae and condylomata acuminata are hyperplasias caused by HPV. Both can clinically mimic SKs but differ in that they most frequently occur in younger individuals. Verrucae often present as rough papules with pinpoint red or black dots that represent dilated or thrombosed capillary loops at the tips of the underlying dermal papillae. Verrucae favor acral locations, while condylomata acuminata occur in the anogenital region and rarely on the buccal mucosa. HPV has been reported in anogenital lesions with histologic features of SK (see above). Koilocytic changes of perinuclear vacuolization with nuclear pyknosis in the superficial epidermis may not be present in some verrucae, but are helpful if present. An SK generally tends to be oriented in a more horizontal than vertical fashion, often has horn pseudocysts, and lacks koilocytes.
Tumor of the follicular infundibulum is distinguished from an SK by a superficial distinct plate-like epithelial proliferation. The latter consists of multiple slender epidermal connections composed of basaloid or pale cells, without as much hyperkeratosis as an SK. The reticulated type of SK has a similar pattern but lacks the distinct plate-like growth. Skin-colored SKs, especially in flexural sites, can easily be mistaken for an acrochordon, which has a pedunculated shape and is usually smoother and smaller in size.
Other conditions with papillated epidermal hyperplasia histologically are readily distinguished on a clinical basis. Some of these include acanthosis nigricans, epidermal nevus, and confluent and reticulated papillomatosis. Epidermal nevi tend to follow the lines of Blaschko, appearing in a linear arrangement on the extremities and a whorled pattern on the trunk. Although histologic sections of epidermal nevi typically exhibit acanthosis and hyperkeratosis, they usually lack the pseudocysts seen in many SKs. Epidermal nevi are also usually apparent at birth or shortly thereafter, unlike SKs, which are seen in adults.
Acrokeratosis verruciformis of Hopf has a “church spire” configuration similar to that described for hyperkeratotic SK , but it is an autosomal dominant disorder and lesions are usually limited to the distal extremities (see Ch. 59 ). Acanthoma fissuratum may clinically resemble a large SK, but histologic analysis reveals irregular epidermal hyperplasia due to friction.
Treatment
Treatment of asymptomatic SKs is largely performed for cosmetic reasons. Symptomatic lesions are usually removed by destruction, curettage or shave excision. The most common method of destruction is cryotherapy. Other methods include electrodesiccation and laser vaporization (e.g. erbium:YAG). Inhibitors of AKT activity have been shown to induce apoptosis in cultured intact seborrheic keratoses and may possibly represent a future topical therapy .
Lichenoid Keratosis
▪ Lichen planus-like keratoses (LPLK) ▪ Solitary lichenoid keratosis (SLK) ▪ Benign lichenoid keratosis (BLK)
- ▪
A pink to red–brown scaly papule that arises in sun-exposed sites, most commonly the forearms and upper chest
- ▪
Histologically, it appears almost identical to lichen planus, but is distinguished by clinical correlation
- ▪
Represents an inflammatory stage of a solar lentigo, SK or actinic keratosis
History
In 1966, two independent groups reported solitary asymptomatic lesions with the histologic features of lichen planus. These two groups referred to these lesions as solitary lichen planus and solitary lichen planus-like keratosis.
Epidemiology
Eighty-five percent of lichenoid keratoses (LKs) develop between the ages of 35 and 65 years . Women are diagnosed with the condition twice as frequently as men, and the vast majority of lesions are seen in Caucasians. Occasionally, patients with a fair complexion and significant photodamage develop multiple LKs.
Pathogenesis
LKs have been thought to represent inflammation of a benign lentigo, SK or actinic keratosis. FGFR3 , PIK3CA and RAS mutations have been detected in ~50% of LKs, providing support for this relationship (see Table 109.1 ) . Because some of the lesions are related to “precancerous” actinic keratoses, the term lichenoid keratosis is preferred over the term benign lichenoid keratosis. Lesions are best referred to as lichenoid actinic keratosis when there is significant keratinocyte cytologic atypia or other classic features of an actinic keratosis, and as irritated SK when a lichenoid infiltrate is associated with other features of an SK.
Increased numbers of Langerhans cells have been observed in the epidermis of LKs. This has led to the suggestion that the lichenoid infiltrate of lymphocytes is secondary to a stimulus from an unidentified epidermal antigen. This mechanism is similar to that proposed for lichen planus.
Clinical Features
LKs occur primarily as solitary pink to red–brown, often scaly, papules ranging from 0.3 to 1.5 cm in diameter ( Fig. 109.5A ). An astute clinician can oftentimes suspect the diagnosis before the biopsy is performed. LK most closely resembles Bowen disease or BCC, and for this reason, these lesions are frequently biopsied. They are usually asymptomatic, although occasionally patients complain of slight pruritus or stinging . The most common sites are the forearm and upper chest, with less frequent occurrence on the shins (in women) and other chronically sun-exposed sites. Dermoscopic findings include light brown pseudonetworks due to residual solar lentigo and overlapping pinkish areas related to lichenoid inflammation. Annular granular structures and gray pseudonetworks are present in the early regressing stage, while blue-gray fine dots can be seen in the late regressing stage. Blue-gray dots or globules, representing melanophages, are also considered typical of an LK.
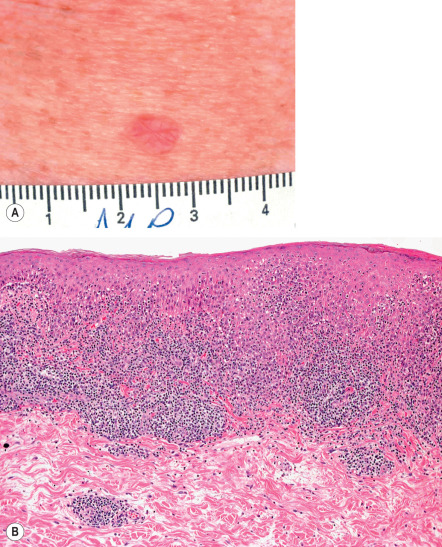
Pathology
LK has a lichenoid infiltrate composed primarily of lymphocytes with scattered histiocytes ( Fig. 109.5B ). Eosinophils and plasma cells are sometimes present. All other elements of an interface dermatitis are observed, including basal vacuolar alteration, melanin incontinence, and colloid bodies. When melanin incontinence is prominent, the term pigmented lichenoid keratosis is sometimes used. Parakeratosis may be seen, unlike in typical lichen planus. Sometimes there is frank separation of the epidermis from the infiltrate in the dermis, giving rise to a subepidermal cleft or blister cavity. Basal cell proliferation is absent, and keratinocyte atypia is either mild or absent. Changes of a solar lentigo or macular SK are often present at the periphery of the specimen.
Occasionally, the histopathologic findings in an LK may mimic mycosis fungoides due to the presence of Pautrier-like microabscesses, alignment of lymphocytes along the basal layer, and epidermotropism of the infiltrate. There is also an atrophic variant, and sometimes the lesion may mimic lupus erythematosus histopathologically .
LKs may undergo regression, and thus, histologically, they may be confused with other regressing tumors, especially melanoma. The regressed LK usually displays a loosely fibrotic papillary dermis with scattered lymphocytes and melanophages. Regressed melanoma typically has a dense band of melanophages with lymphocytes and dilated blood vessels; there is also usually a thin epidermis overlying effaced rete ridges. In states of partial regression, histologic clues to the proper diagnosis remain. The end stages of both tumors may not be distinguishable. Careful search for elements of superficial BCC is also advisable.
Differential Diagnosis
Frequently cited clinical impressions include Bowen disease, actinic keratosis, BCC, irritated SK, melanoma (including amelanotic), and melanocytic nevus. Development of inflammation or color change within a pigmented lesion may lead the clinician to suspect an atypical melanocytic nevus or melanoma . Even when a melanocytic neoplasm is not in the clinical differential diagnosis, the pathologist must take care to ensure that the lichenoid infiltrate is not obscuring melanoma in situ or even melanoma involving the superficial dermis, as the latter may become heavily inflamed. Multiple LKs may be misdiagnosed as a lichenoid drug eruption and can be a cause of pruritus in elderly patients .
Histologically, LK resembles other conditions with a band-like inflammatory (lichenoid) reaction pattern, including lichen planus, “lichenoid” lupus erythematosus, and lichenoid fixed drug reactions. Clinical correlation is very useful in excluding these conditions, e.g. LK occurs as a single lesion in the vast majority of patients. While lichen planus may be virtually identical to LK histologically, there is usually more wedge-shaped hypergranulosis and less parakeratosis in lichen planus.
Treatment
Once the diagnosis of LK is made, no further therapy is necessary. Any remaining lesion may be destroyed by any method.
Dermatosis Papulosa Nigra
- ▪
Most common in individuals of African descent with darkly pigmented skin
- ▪
Multiple hyperpigmented papules on the face
- ▪
Considered to be a variant of SK
- ▪
Cryotherapy can result in hypopigmentation; optimal treatment is scissor snip, curettage or electrodesiccation, when desired
Introduction
Dermatosis papulosa nigra (DPN) presents as multiple, hyperpigmented, filiform to sessile, smooth-surfaced papules measuring from 1 to 5 mm. The lesions typically develop on the face of darkly pigmented individuals ( Fig. 109.6 ).
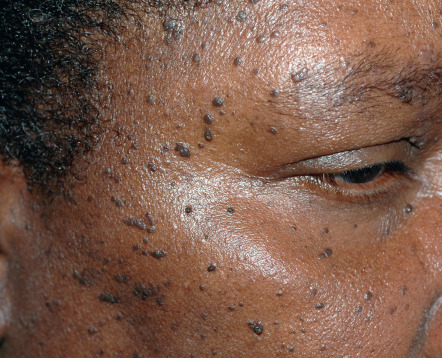
History
DPN was first described by Castellani in 1925 based on his observations during visits to Central America and Jamaica.
Epidemiology
DPN has a strong familial predisposition and in selected populations, reported incidences in selected study populations have ranged from 10% to 75%. This variability in incidence may be a reflection of sampling, as more lightly pigmented African-Americans are affected less commonly . In addition to those with African heritage, the condition has also been reported in Filipinos, Vietnamese, Europeans and Mexicans . Women are twice as likely to be affected as men and rarely it occurs in children. DPN is not related to any systemic disease or syndrome.
Pathogenesis
The cause of DPN is unknown. It tends to have an earlier age of onset than that of SKs, but otherwise is similar and probably is a variant of SK. Some authors view it as a variant of multiple acrochordons. However, an FGFR3 mutation was detected in two samples of DPN, supporting its relationship to SKs (see Table 109.1 ) .
Clinical Features
Pigmented papules are distributed symmetrically on the malar eminences and forehead. Less often, lesions are on the neck, chest and back. The papules usually appear during adolescence and gradually increase in size and number over time, peaking in the sixth decade.
Pathology
DPN is characterized by acanthosis, papillomatosis and hyperkeratosis in a pattern quite similar to the acanthotic type of SK. Horn pseudocysts, however, are not a common feature.
Differential Diagnosis
The differential diagnosis of DPN includes primarily multiple SKs and acrochordons. Occasionally, verrucae, melanocytic nevi, adnexal tumors (e.g. trichoepitheliomas, fibrofolliculomas, tricholemmomas, basaloid follicular hamartomas, syringomas), and angiofibromas might be considered clinically (see Fig. 111.5 ).
Treatment
Treatment of DPN is generally performed only for cosmetic purposes, although individual lesions may be troublesome. Care should be exercised to avoid treatments that may result in dyspigmentation, so initially it may be best to treat just a small number of lesions. Snip excision with scissors, curettage, and light electrodesiccation are the most common treatment modalities. Hypopigmentation after cryotherapy can be particularly problematic, since melanocytes are more sensitive to freeze damage than are keratinocytes.
Stucco Keratosis
▪ Keratosis alba
- ▪
Gray–white papules
- ▪
Typically on the lower extremities of older adults, especially the ankles
- ▪
Lesions are “stuck on”, and when scraped off, there is minimal bleeding
- ▪
Probably a variant of SK
Introduction
Stucco keratoses are discrete, gray–white papules that are hard and opaque, with the latter reflecting focal accumulations of cornified material. They are usually distributed symmetrically below the knee, especially on the ankle and dorsal aspect of the foot.
History
In 1958, Kocsard was the first to describe stucco keratoses and eight years later he coined the term stucco keratosis to reflect its “stuck-on” appearance. Although early accounts of stucco keratoses described elastotic changes in the upper dermis, this has been shown to represent solar elastosis.
Epidemiology
There is no familial predilection for stucco keratoses. They can become more apparent during cold winter months, most likely due to lower humidity and drier skin. They occur primarily in middle-aged to elderly individuals and are usually first observed after 40 years of age. Men are four times more likely to be affected than women. Although precise epidemiologic data are lacking, most descriptions have been in Caucasians. There are no known associated diseases or syndromes.
Pathogenesis
Histochemical studies suggest an acquired focal abnormality of keratinization, and a PIK3CA mutation was detected in three of five samples of stucco keratoses . There was a single case report of multiple HPV types within various keratoses detected by PCR, but the significance of this finding is unclear as HPV DNA is purported to be detectable in at least 20% of normal skin specimens . Ultrastructural examination has revealed no viral particles .
Sun exposure has been proposed to be a factor in the formation of stucco keratoses, as most affected individuals have evidence of solar damage. However, this may be a reflection of the age and phototype of the patients. Heat and petroleum products such as tar have also been implicated, but none of these theories explain the distribution pattern.
Clinical Features
Usually tens, but sometimes hundreds, of gray–white papules are present on the lower extremities of older adults ( Fig. 109.7 ). They favor the ankles and dorsal feet but may extend to the thighs. Occasionally, stucco keratoses appear on the forearms. The lesions are usually small, measuring from 1 to 4 mm; rarely larger plaques are seen. The lesions may be scraped or flicked off the skin surface with a fingernail and there is usually minimal, if any, bleeding. A collarette of dry scale may remain. On occasion, brown, pink and deep yellow shades have been described.
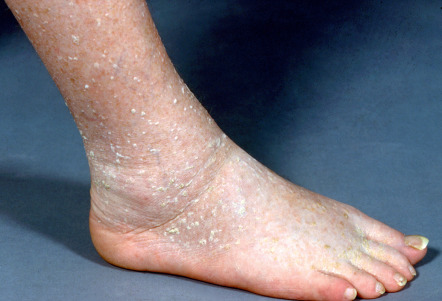
Pathology
Stucco keratoses display prominent orthokeratotic hyperkeratosis and papillomatosis, often imparting a peaked “church spire” pattern. Acanthosis is usually present, but to a lesser degree than in some SKs. The granular layer may be thickened. Horn cysts are usually absent and cytologic atypia is not a feature.
Differential Diagnosis
Stucco keratoses may resemble SKs, acrokeratosis verruciformis of Hopf, verruca plana, or epidermodysplasia verruciformis. SKs tend to be larger and pigmented with a surface that appears “greasy”, in contrast to stucco keratoses, which are smaller and have a dry, rough surface. Histologically, stucco keratoses resemble hyperkeratotic SKs and acrokeratosis verruciformis; all three may demonstrate papillomatosis and digitation, giving an appearance similar to “church spires”. As discussed below, the lesions of acrokeratosis verruciformis favor the dorsal aspects of the hands (as well as the feet).
Treatment
As with SKs, treatment is generally for cosmetic reasons, although symptomatic lesions may require removal. Curettage, or local destruction with cryosurgery or electrodesiccation, are the usual forms of treatment. Topical application of urea, lactic acid, or retinoids may reduce the hard scale, but the actual lesions persist.
Porokeratosis
- ▪
At least six clinical variants of porokeratosis are recognized
- ▪
The prototype, porokeratosis of Mibelli , is a plaque that appears during infancy or childhood, usually on an extremity
- ▪
Disseminated superficial actinic porokeratosis (DSAP) is the most common type, with multiple thin papules appearing in chronically sun-exposed skin of the extremities
- ▪
Linear porokeratosis develops during infancy or childhood and follows the lines of Blaschko
- ▪
Punctate porokeratosis appears during or after adolescence as 1–2 mm papules of the palms and soles
- ▪
Porokeratosis palmaris et plantaris disseminata (PPPD) is a variation of punctate porokeratosis, with lesions also present on other areas of the body
- ▪
Porokeratosis ptychotropica is a rare variant that favors the intergluteal cleft and buttocks
- ▪
Some forms are inherited in an autosomal dominant fashion (e.g. DSAP)
- ▪
A thread-like raised hyperkeratotic border is characteristic and its histologic correlate is the cornoid lamella, a thin column of parakeratosis
- ▪
Development of SCC is possible but rare
Introduction
Porokeratosis presents as a keratotic papule or plaque, often with an annular appearance due to its thread-like elevated border that expands centrifugally. The disorder was erroneously named porokeratosis because the column of parakeratosis, known as the cornoid lamella, was initially described as being present over a sweat pore, which of course is a fixed structure that cannot expand peripherally. At least six types of porokeratosis have been recognized ( Table 109.2 ). There are reports of more than one type of porokeratosis developing in the same patient and in multiple members of an affected family .
| CLINICAL VARIANTS OF POROKERATOSIS | |
|---|---|
| Variant | Clinical features |
| Porokeratosis of Mibelli | Plaque that arises during infancy or childhood, usually on a distal extremity; often several cm in diameter |
| Disseminated superficial actinic porokeratosis (DSAP) | Pink to brown papules and plaques with peripheral scale; arise in sun-exposed sites, especially the extensor forearms and shins; usually measure from a few mm to 1 cm in diameter; in some patients, autosomal dominant inheritance |
| Linear porokeratosis | Streaks along the lines of Blaschko; arise during infancy or childhood; risk of development of squamous cell carcinoma |
| Punctate porokeratosis | Palmoplantar papules that measure 1–2 mm in diameter; arise during adolescence or adulthood |
| Porokeratosis palmaris et plantaris et disseminata (PPPD) | Palmoplantar papules in addition to involvement of the trunk, extremities, and even mucous membranes; onset during childhood or adolescence |
| Porokeratosis ptychotropica | Red to brown papules or plaques in the intergluteal cleft and on the buttocks; there may be coalescence of lesions centrally with scattered papules peripherally |
| Eruptive disseminated porokeratosis | Abrupt onset of numerous, widespread, inflamed keratoses; may be a sign of an associated malignancy in up to ~30% of patients |
History
In 1893, Mibelli first described what is now considered classic porokeratosis: one to several discrete plaques that may occur anywhere on the skin or mucous membranes, usually appearing during infancy or childhood. That same year, Respighi reported a superficial and disseminated form of the disease while Chernosky defined disseminated superficial actinic porokeratosis (DSAP) nearly 70 years later. In the 1970s, porokeratosis palmaris et plantaris disseminata (PPPD) and punctate porokeratosis were described. Porokeratosis ptychotropica is the most recently recognized form.
Epidemiology
Porokeratosis can be inherited as an autosomal dominant disorder, including DSAP and PPPD, but many cases appear to be sporadic. Porokeratosis of Mibelli affects boys more than girls, but DSAP is more common in women. Although PPPD is inherited in an autosomal dominant manner, it is twice as common in males than in females, perhaps reflecting an environmental factor. Given its relationship to sun exposure, DSAP is more common in Caucasians and is rare in blacks. Linear porokeratosis has been reported in monozygotic twins , and it has been seen in families with other types of porokeratosis (see below) .
Pathogenesis
Although porokeratosis is thought to be a disorder of keratinization, the definitive pathogenesis remains unclear. There are multiple genetic loci for porokeratosis , including 12q24 (POROK2; PPPD), 12q24 (POROK3; DSAP1; mutation in MVK ), 15q25–q26 (POROK4; DSAP2) , 1p31 (POROK5; DSAP3) , 18p11 (POROK6; DSAP4), 16q24 (POROK7; DSAP; mutation in MVD ), 20q13 (POROK8; DSAP; mutation in SLC17A9 ), and 1q22 (POROK9; DSAP; mutation in FDPS ). Of note, POROK1 initially referred to porokeratosis of Mibelli, but now includes other variants such as linear porokeratosis, and it is associated with mutations in PMVK which encodes phosphomevalonate kinase. MVK encodes mevalonate kinase, which may play a role in calcium-induced keratinocyte differentiation and proliferation, and SLC17A9 encodes a vesicular nucleotide transporter . There is ongoing debate regarding two other genes: SSH1 and SART3 .
An older hypothesis set forth by Reed proposes that lesions of porokeratosis represent an expanding mutant clone of keratinocytes. In Reed’s view, the characteristic cornoid lamella, seen histologically, represents the border between normal epidermis and the mutant clone of cells. A dermal lymphocytic infiltrate beneath the cornoid lamella or in the central zone of the lesion is considered to represent an immunologic response. Supporting this theory is the finding of abnormal DNA ploidy in keratinocytes of porokeratosis. Triggering factors such as UVR exposure or immunosuppression due to AIDS or organ transplantation may induce porokeratosis in individuals who are genetically predisposed to developing abnormal clones of keratinocytes. A second, less accepted theory suggests that the dermal lymphocytic infiltrate may be directed against an unidentified epidermal antigen and that this population of inflammatory cells releases mediators that provide a mitotic stimulus for epidermal cells .
Clinical Features
Classic porokeratosis of Mibelli is a rare condition, appearing during infancy or childhood as an asymptomatic, small, brown to skin-colored keratotic papule that gradually enlarges over a period of years to form a plaque that may measure several centimeters in diameter. There is a raised, sharply demarcated, keratotic, thready border with a longitudinal furrow ( Fig. 109.8A ). The center of the lesion may be hyperpigmented, hypopigmented, atrophic, and/or anhidrotic. Lesions may occur anywhere on the body, including mucous membranes , but the extremities are most frequently involved. Multiple lesions may be present, but they are almost always regionally localized and unilateral.