Abstract
The cutaneous amyloidoses represent a heterogeneous group of conditions in which amyloid, a fibrillar material that can result from the degradation of various proteins, is deposited in the skin. In primary cutaneous amyloidosis, the deposits are derived from keratin (macular, lichen, biphasic) or immunoglobulin light chains (nodular). The specific cutaneous lesions of primary systemic amyloidosis are waxy, translucent or purpuric papules, nodules and plaques. Primary systemic amyloidosis is due to a plasma cell dyscrasia while secondary systemic amyloidosis arises from chronic inflammatory conditions such as rheumatoid arthritis or in the setting of chronic infections. Treatment of all forms of amyloidosis is challenging, and although the primary cutaneous forms are not life-threatening, primary systemic amyloidosis can carry a poor prognosis such that early recognition and appropriate management are vital.
Keywords
amyloid, Congo red stain, Thioflavin T stain, amyloidosis, primary cutaneous amyloidosis, secondary cutaneous amyloidosis, macular amyloidosis, lichen amyloidosis, lichen amyloidosus, biphasic amyloidosis, nodular amyloidosis, secondary systemic amyloidosis, primary systemic amyloidosis
- ▪
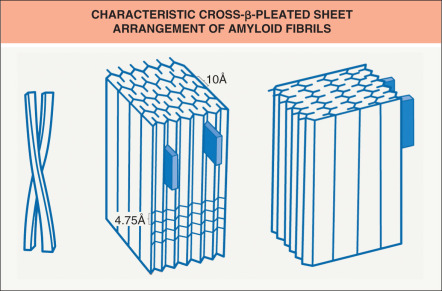
Characteristic properties of amyloid include congophilia and green birefringence under polarized light, a distinctive fibrillar ultrastructure, and a cross-β-pleated sheet configuration by X-ray crystallography
- ▪
The three major forms of primary cutaneous amyloidosis are:
- •
macular amyloidosis: confluent or rippled hyperpigmentation, often on the back and the upper arm
- •
lichen amyloidosis: hyperpigmented papules, often in a rippled pattern; favors the extensor surfaces of the extremities and back
- •
nodular amyloidosis: waxy, skin-colored to pink–yellow nodules
- •
- ▪
Biphasic amyloidosis has features of both macular amyloidosis and lichen amyloidosis
- ▪
The amyloid deposits in macular amyloidosis and lichen amyloidosis are keratinocyte-derived, whereas those in nodular amyloidosis are composed of immunoglobulin light chains and are often accompanied by an infiltrate of plasma cells
- ▪
Mucocutaneous lesions are seen in 30–40% of patients with primary systemic (AL) amyloidosis and include waxy papulonodules and plaques, ecchymoses, pinch purpura, and macroglossia; carpal tunnel syndrome in association with the latter is a classic presentation
- ▪
In the absence of specific mucocutaneous lesions, aspiration of abdominal fat can be performed as amyloid deposits may be detected in many patients with primary systemic amyloidosis
- ▪
Once the diagnosis of AL amyloidosis is established, evaluation for systemic involvement is indicated
Introduction
Amyloidosis is not a single disease; rather, the term is used to refer to several diseases that share the common feature of abnormal extracellular deposition of amyloid, a fibrillar proteinaceous material, within tissues . Deposits of amyloid can be seen in a range of clinical disorders, from plasma cell dyscrasias and Alzheimer disease to familial polyneuropathies and primary cutaneous lichen amyloidosis.
Amyloid itself is not a single chemically distinct substance, and several types of amyloid have been described. However, regardless of the source, pathogenetic mechanisms, or underlying disease, amyloid material shares certain common tinctorial and physico-chemical properties, e.g. the cross-β-pleated sheet configuration .
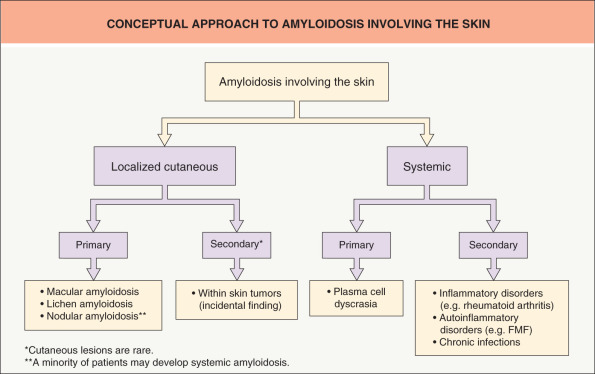
The two clinical settings in which dermatologists are likely to encounter amyloidoses are: (1) the more common primary cutaneous forms of amyloidosis; and (2) the less common systemic amyloidosis with cutaneous manifestations ( Fig. 47.1 ).
History
Virchow, in 1854, introduced the term “amyloid” . He believed that the substance resembled starch or cellulose because, like starch, it turned blue when stained with iodine followed by dilute sulfuric acid. In 1928, Gutmann first described a patient with clinical features of lichen amyloidosis, while Freudenthal, in 1930, introduced the term “lichen amyloidosus” .
Epidemiology
The precise epidemiology of systemic amyloidosis is not known, as the disease is often underdiagnosed or misdiagnosed . In the US, the incidence of primary systemic amyloidosis is estimated to be ~1275 to 3200 new cases per year ; an increase in detection will likely accompany more widespread use of the assay for serum free light chains. Since 1980, there has been a marked decline in the incidence of symptomatic rheumatoid arthritis-associated secondary systemic amyloidosis due to better control of the inflammatory response .
Primary cutaneous amyloidosis is commonly observed in Southeast Asian countries, including Singapore, Taiwan, and Thailand . Lichen amyloidosis appears to be more common in those of Chinese descent. Macular amyloidosis is also commonly seen in Central and South American countries, especially those close to the equator . In general, macular amyloidosis and lichen amyloidosis occur more frequently in individuals with skin phototypes III and IV.
Classification
Amyloidosis can be classified clinically into systemic (generalized) forms, with involvement of several organ systems, and organ-limited (localized) forms, in which deposits are limited to a single organ such as the skin ( Table 47.1 ). In the localized forms, amyloid deposition occurs at or near the site of synthesis, while in the systemic forms the precursors are secreted into the circulation, followed by amyloid deposition at distant sites . Amyloidosis can also be classified according to its constituent precursor protein ( Table 47.2 ; see next section).
| CLINICAL CLASSIFICATION OF AMYLOIDOSIS |
| Organ-limited (localized) amyloidosis |
Cutaneous
Cerebral
|
| Systemic amyloidosis |
Primary systemic amyloidosis (AL ≫ AH amyloid protein)
Secondary systemic amyloidosis (reactive; AA amyloid protein)
ATTR amyloidosis (transthyretin amyloid protein ** )
ALect2 amyloidosis (leukocyte chemotactic factor 2) See Table 47.2 for additional forms |
* Seen in patients with Sipple syndrome.
** Transthyretin is a serum trans port protein for thy roxine and retin ol-binding protein that is produced in the liver, choroid plexus and eye ; orthotopic liver transplantation represents an effective treatment for the inherited forms of ATTR amyloidosis .
| CHEMICAL CLASSIFICATION OF AMYLOIDOSES | ||
|---|---|---|
| Precursor protein | Amyloid protein | Clinical syndrome |
| Aβ precursor protein (AβPP) | Aβ | Alzheimer disease, aging |
| (Apo) serum AA * | AA | Secondary systemic amyloidosis (see Table 47.1 ), hereditary periodic fever syndromes (e.g. familial Mediterranean fever, Muckle–Wells syndrome, TRAPS; see Ch. 45 ) |
| Apolipoprotein AI | AApoAI | Hereditary apolipoprotein AI-associated amyloidosis; distribution of organ involvement, including skin, related to location of mutation |
| Apolipoprotein AII | AApoAII | Familial renal amyloidosis |
| β 2 -microglobulin | Aβ 2 M | Chronic hemodialysis |
| Calcitonin | ACal | Medullary carcinoma of the thyroid |
| Cystatin C | ACys | Hereditary cystatin C amyloid angiopathy † |
| Gelsolin | AGel | Familial amyloidosis, Finnish type |
| Immunoglobulin heavy chain (very rare) ‡ | AH | Primary systemic amyloidosis |
| Keratin | AKer | Primary (localized) cutaneous amyloidosis |
| Immunoglobulin light chain | AL | Primary systemic amyloidosis (associated with plasma cell dyscrasia § ≫ multiple myeloma), primary cutaneous nodular amyloidosis |
| Insulin | AIns | Firm nodule at sites of repeated insulin injections |
| Leukocyte chemotactic factor 2 | ALect2 | Progressive renal insufficiency and hepatic involvement; favors Mexican-Americans |
| Transthyretin | ATTR | ATTR amyloidosis: (1) familial amyloid polyneuropathy ( TTR mutations); (2) familial amyloid cardiomyopathy ( TTR mutations); and (3) wild-type ATTR amyloidosis/senile systemic amyloidosis/senile cardiac amyloidosis |
| Corneodesmosin | – | Hypotrichosis simplex of the scalp |
* Amyloid-associated protein synthesized by the liver.
† Detection of deposits within clinically normal skin allows diagnosis prior to clinical symptoms (cerebral hemorrhage).
‡ More commonly, heavy chains produce a non-amyloid type deposit termed heavy chain deposition disease which is sometimes associated with acquired cutis laxa.
§ Monoclonal gammopathy; progression depends upon risk factors (e.g. non-IgG paraprotein).
Pathogenesis
The major component of amyloid is the fibril protein; the minor components are amyloid P component, glycosaminoglycans, and apoE lipoprotein. Amyloid P is a glycoprotein derived from serum amyloid P protein (SAP) and it has a specific calcium-dependent binding affinity for amyloid. Approximately 30 distinct forms of amyloid fibril proteins and their precursors have been identified, including: AL (amyloid light chains), containing immunoglobulin light chains; AA (amyloid-associated), composed of an acute phase protein synthesized by the liver; Aβ amyloid, found in cerebral lesions of Alzheimer disease; and ATTR (transthyretin-associated), found in some forms of familial amyloidosis (mutated) and senile systemic amyloidosis (wild-type) . Each condition is associated with a specific precursor protein ; these amyloid precursors are initially soluble proteins that undergo changes leading to aggregation, polymerization, fibril formation, and, finally, extracellular tissue deposition as insoluble amyloid.
The process by which this transformation takes place differs amongst the various types of amyloidoses. In primary systemic amyloidosis, substitution of amino acids at specific positions within the variable region of the immunoglobulin light chain potentially destabilizes these chains thereby increasing the likelihood of their conversion to amyloid fibrils. Similarly, mutations in the transthyretin gene have been shown to significantly alter the stability of the transthyretin protein and increase its baseline mild amyloidogenicity . The accumulation of these relatively inert amyloid fibrils within vital organs leads to pressure atrophy and functional impairment.
The precise pathogenesis of primary cutaneous amyloidosis is not yet fully understood. Prolonged friction, genetic predisposition, Epstein–Barr virus, and environmental factors have all been proposed as possible etiologic factors. The precursor protein involved has not been fully characterized; however, for the macular and lichenoid variants of primary cutaneous amyloidosis it is thought to be keratinocyte-derived. This has been supported by ultrastructural studies demonstrating transitional forms between viable keratinocytes and amyloid, as well as by positive reactions with monoclonal antibodies directed against basal layer keratins. The fibrillar theory proposes that keratinocyte tonofilaments undergo degeneration and pass into the dermis, where they are presumably modified by histiocytes and fibroblasts into amyloid material. An alternative theory suggests that the material is produced at the epidermo-dermal interface, with precursor proteins being secreted by basal keratinocytes. This hypothesis has been supported by ultrastructural findings and demonstration of basement membrane antigens such as type IV collagen and laminin within amyloid deposits . Although these cutaneous amyloid deposits stain positively with anti-human antibodies directed against IgG, IgM and IgA, this staining is thought to be the result of nonspecific immunoglobulin absorption as opposed to an immunoglobulin being the putative precursor protein. Apolipoprotein E4, galectin-7, and actin have also been shown to be associated with primary cutaneous amyloid deposits and may be synthesized locally by epidermal keratinocytes . Small fiber neuropathy has been observed in primary cutaneous amyloidosis and may be related to the associated pruritus .
In nodular amyloidosis there is no specific staining with antikeratin antibodies, but rather the amyloid deposits are composed of immunoglobulin light chains, suggesting plasma cell derivation akin to the cutaneous lesions of primary systemic amyloidosis. Thus, its origin is very different from that of either macular or lichen amyloidosis . Presumably, there is local cutaneous production of light chains in nodular amyloidosis, whereas in primary systemic amyloidosis with skin involvement, the light chains are derived from the systemic circulation.
Amyloid properties
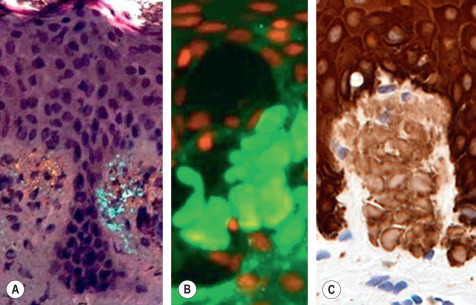
In H&E-stained sections, amyloid appears as amorphous, eosinophilic fissured masses ( Fig. 47.2 & Table 47.3 ). With Congo red, amyloid has an orange–red color on routine light microscopy, whereas under polarized light it exhibits green birefringence ( Fig. 47.3A ). Other special stains that can be used to detect amyloid deposits include crystal violet, methyl violet, periodic acid Schiff (PAS), Sirius red, pagoda red, Dylon stain, and Thioflavin T ( Fig. 47.3B ) . AA (but not AL) amyloid loses its affinity for Congo red after exposure to potassium permanganate.
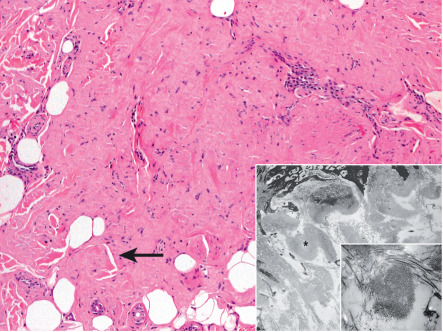
| Histologic criteria used to define amyloid |
|---|
|
By electron microscopy, amyloid appears as 7 to 10 nm wide, non-branching, non-anastomosing fibrils (see Fig. 47.2 ). X-ray crystallography and infrared spectroscopy reveal a characteristic cross-β-pleated sheet conformation ( Fig. 47.4 ). These findings are identical in all types of amyloid, regardless of clinical setting or chemical composition . The cross-β-pleated sheet structure of amyloid is believed to be responsible for its staining and birefringence with Congo red.