Abstract
Adnexal neoplasms encompass both benign and malignant tumors with differentiation towards hair follicles as well as the ducts and secretory components of sebaceous, eccrine and apocrine glands. Those tumors with follicular differentiation can be further divided based upon the type of keratinization they display, e.g. infundibular, isthmic, germinative, matrical, or resembling the inner and outer root sheath. Because many of the adnexal neoplasms have ductal differentiation and eccrine ducts can be difficult to distinguish from apocrine ducts, it is often impossible to determine if a neoplasm is eccrine or apocrine in origin. In fact, many adnexal tumors previously considered to be of eccrine differentiation have been reclassified into the apocrine group. Most adnexal neoplasms are benign, but malignant variants of virtually every entity can be encountered, sometimes with deceptively mild morphologic features. Diagnosis and classification are based on accurate evaluation of clinicopathologic features.
Keywords
adnexal neoplasms, adnexal tumors, hair follicle nevus, trichofolliculoma, fibrofolliculoma, perifollicular fibroma, trichodiscoma, nevus sebaceus, mixed tumor, chondroid syringoma, trichoblastoma, trichoepithelioma, desmoplastic trichoblastoma, desmoplastic trichoepithelioma, pilomatricoma, pilomatrical carcinoma, tricholemmoma, trichilemmoma, tumor of follicular infundibulum, infundibuloma, isthmicoma, trichoadenoma, sebaceous gland hyperplasia, sebaceous adenoma, sebaceoma, sebaceous epithelioma, sebaceous carcinoma, poroma
- ▪
Terminology and classification of adnexal tumors has varied considerably according to different authorities
- ▪
Adnexal neoplasms can differentiate toward the folliculosebaceous-apocrine unit or the eccrine apparatus
- ▪
As a reflection of joint ontogeny, tumors often exhibit combinations of follicular, sebaceous and apocrine differentiation
- ▪
Many adnexal tumors historically classified as eccrine are probably apocrine
The nosology of adnexal neoplasms has been confused for decades, and a key problem has been the lack of a logical classification scheme. Postulated classifications and inferences regarding lineage have often been contradictory, a reflection of the fact that broad conclusions regarding lineage and classification were formerly based on enzyme histochemical attributes of dubious specificity .
The crucial principle to keep in mind when considering adnexal neoplasms is that the development of the eccrine apparatus is distinct from the folliculosebaceous-apocrine unit. Eccrine glands develop directly from embryonic epidermal anlage during early months of fetal development . Follicles arise directly from the epidermis concurrently, but their development differs in that mesenchymal cells, precursors of the follicular papilla, descend jointly into the dermis with the developing follicle (See Ch. 2 and Fig. 2.4 ). Subsequently, sebaceous and apocrine glands and their ducts elaborate as secondary structures from bulges on the side of the developing follicle.
These ontogenetic relationships reflect relationships observed repeatedly in clinical disease . As one would expect from ontogeny, follicular, sebaceous and apocrine differentiation occur conjointly, and combinations of eccrine and folliculosebaceous differentiation probably do not exist.
Both the differentiation of the tumor ( Fig. 111.1 ) and the topography of adnexal structures offer insights into logical classification. There is striking variation in anatomic distribution among adnexal neoplasms, and some of these differences hold implications regarding lineage. Based on topography, it is nonsensical to pigeonhole spiradenoma, a tumor that commonly occurs within apocrine skin and rarely, if ever, develops within acral skin, as an eccrine tumor. In contrast, an acral predilection suggests that poroma is commonly of eccrine lineage .
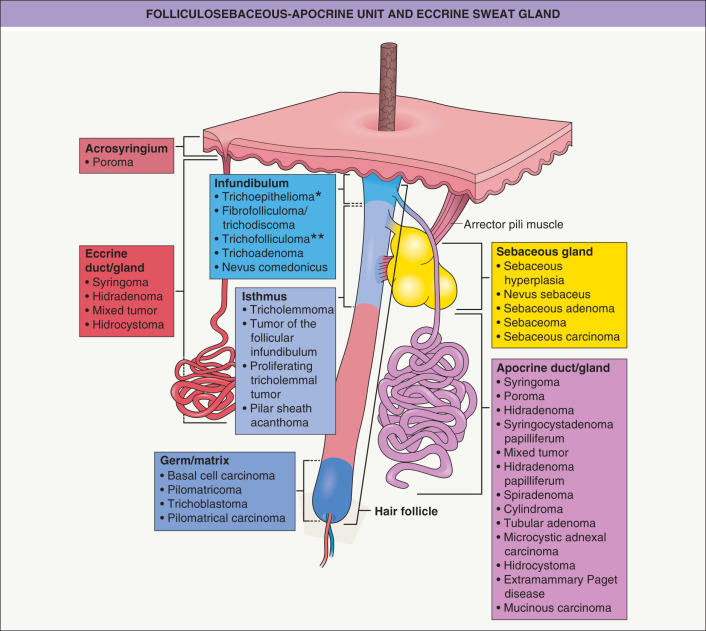
Lastly, microscopy and morphology play a role in the assessment of lineage. For some lines of differentiation, the meaning ascribed to specific attributes is indisputable. Cells with coarsely vacuolated cytoplasm and scalloped nuclei signify sebaceous differentiation. Follicular differentiation is established by the presence of bulbar basaloid cells accompanied by mesenchymal cells resembling the follicular papilla. Follicular lineage is also connoted by attributes such as matrical or outer sheath differentiation. In contrast, there are no specific attributes that permit recognition of eccrine or apocrine differentiation, although decapitation configuration at the luminal border can be considered suggestive of apocrine differentiation.
Neoplasms and Proliferations with Follicular Differentiation
Follicular and Folliculosebaceous-Apocrine Hamartomas
Hamartomas are benign proliferations composed of cellular elements, native to a site, in aberrant proportion. A congenital hamartoma, such as nevus sebaceus, is properly referred to as a nevus . Hamartomas can be acquired and present in a tumor-like fashion, although such lesions do not represent authentic neoplasms.
Proliferations of the folliculosebaceous-apocrine unit are commonly hamartoma-like. The folliculosebaceous-apocrine unit incorporates follicular epithelial, follicular mesenchymal, and sebaceous and apocrine elements; thus its microscopically diverse progeny is not surprising. Many follicular proliferations that we consider neoplastic, such as trichoblastoma, have integral stroma, and thus the distinction between follicular hamartoma and follicular neoplasm is sometimes blurred.
Hair follicle nevus
Introduction
Hair follicle nevi are authentic hamartomas, usually congenital, in which hairs and the follicles that ensheath them have abnormal morphology or size or are present in increased numbers.
Clinical features
Hair follicle nevus, also known as vellus hamartoma , presents as a small papule from which fine hairs protrude evenly from the surface. Lesions typically involve the face and commonly reside near the ear . Accessory tragus overlaps with hair follicle nevus, and it has been suggested that the two entities are the same . Nevus pilosus designates hamartomas characterized by closely set, non-vellus hairs.
Pathology
Microscopically, hair follicle nevi display a domed surface with an increase in normally formed vellus follicles. The configuration may resemble normal skin. Accessory tragi exhibit an identical pattern but are uniquely identifiable if deep subjacent hyaline cartilage is present and can be found. Nevus pilosus is characterized by closely set terminal follicles.
Treatment
No therapy is required. Excision can be considered for cosmesis. In the evaluation of a hair follicle nevus or accessory tragus in a small child, parents can be counseled that the relative size of the lesion will diminish with time. The child will grow disproportionately faster than the lesion.
Trichofolliculoma
Introduction
Trichofolliculoma does not represent an authentic neoplasm. Rather, the term designates a group of follicular hamartomas in which fully formed follicular structures emanate from a central dilated infundibular space.
Clinical features
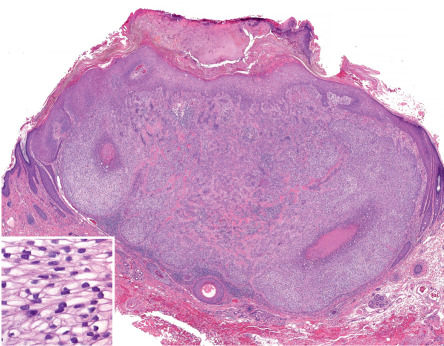
Trichofolliculoma presents as a papule or nodule involving the face, scalp or upper trunk. Many examples are not clinically distinctive. Sometimes, a central follicular ostium or punctum may be identifiable and it may harbor a small tuft of more lightly pigmented hairs ( Fig. 111.2 ). Rarely, the clinical presentation will be as a large nodule or cyst or as multiple lesions in uncommon sites.
Pathology
Prototypically, a trichofolliculoma consists of a central cystic space with infundibular cornification and central orthokeratin. Sometimes, cross-sections of hair shafts are identifiable within the cyst. Relatively well-developed and occasionally oddly formed vellus follicles protrude in radial fashion from the central structure ( Fig. 111.3 ). The follicles usually display a bulb and papilla and exhibit inner and outer sheath and isthmic differentiation. The entire structure, including the central cyst and its associated radiating follicles, is enveloped by a vascularized fibrous (angiofibroma-like) stroma.
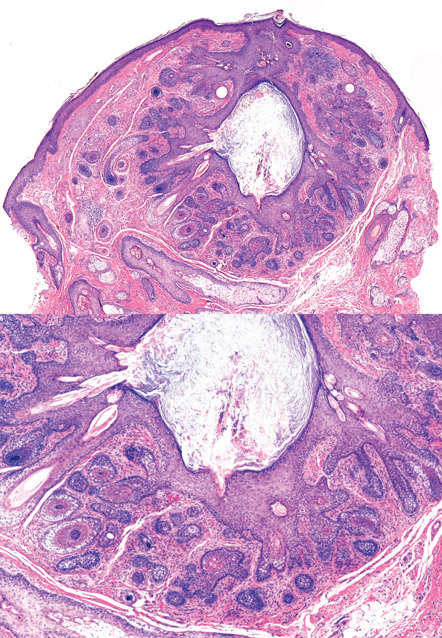
The radiating follicles are frequently cut on tangent, thus creating considerable microscopic variation, and deeper sections through the tissue block sometimes are required for diagnosis. If the radiating follicles are accompanied by sebaceous glands, the hamartoma can be termed a sebaceous trichofolliculoma . This diagnostic embellishment is not of any clinical import.
The term folliculosebaceous cystic hamartoma has been applied to a group of nodular and cystic lesions described independently from trichofolliculoma . Folliculosebaceous cystic hamartoma shows a central infundibular cyst from which follicular and sebaceous elements emanate, and the encompassing stroma may be fibrous or myxoid and often contains adipocytes. Rather than a unique entity, is seems likely that folliculosebaceous cystic hamartoma represents a large cystic sebaceous trichofolliculoma .
Pilar sheath acanthoma is characterized by a cystically dilated follicular configuration that opens to the surface epithelium, much like trichofolliculoma. However, in pilar sheath acanthoma, the lobules of cells that radiate from the cystic dilation recapitulate only the follicular isthmus and outer sheath rather than forming full follicular structures. In addition, a pilar sheath acanthoma does not have the investing fibrous stroma of a trichofolliculoma.
Treatment
Trichofolliculoma is wholly benign and no treatment is needed. If a trichofolliculoma is discovered by biopsy, no further intervention is required.
Fibrofolliculoma, perifollicular fibroma, and trichodiscoma
Introduction
Fibrofolliculoma represents an adnexal hamartoma that includes both follicular epithelial and mesenchymal elements. Perifollicular fibroma and trichodiscoma were described independently but probably represent purely mesenchymal proliferations on the same spectrum as fibrofolliculoma. Some authorities consider perifollicular fibroma to be related to angiofibroma.
Clinical features
Fibrofolliculomas ( Fig. 111.4A,B ), perifollicular fibromas, and trichodiscomas are not clinically distinctive. All present as small, skin-colored to hypopigmented papules that involve the head and neck or upper trunk. The papules commonly present in multiplicity. In this context, strong consideration should be given to the possibility of the Birt–Hogg–Dubé syndrome ( Fig. 111.5 ), an autosomal dominant disorder typified by multiple fibrofolliculomas, trichodiscomas, and acrochordon-like lesions. The major systemic manifestations are renal tumors, the majority of which are oncocytomas or renal cell carcinomas of the chromophobe or hybrid chromophobe–oncocytoma type, pulmonary cysts, and spontaneous pneumothoraces ( Table 111.1 ). Colonic polyps and connective tissue nevi have also been reported in affected patients as have multiple primary cutaneous melanomas . However, it has not yet been determined if there is a significant increased risk for melanoma compared to the general population. Heterozygous mutations in the tumor suppressor gene FLCN , which encodes folliculin, a protein involved in cell–cell adhesion, underlie this syndrome (see Table 111.1 ).
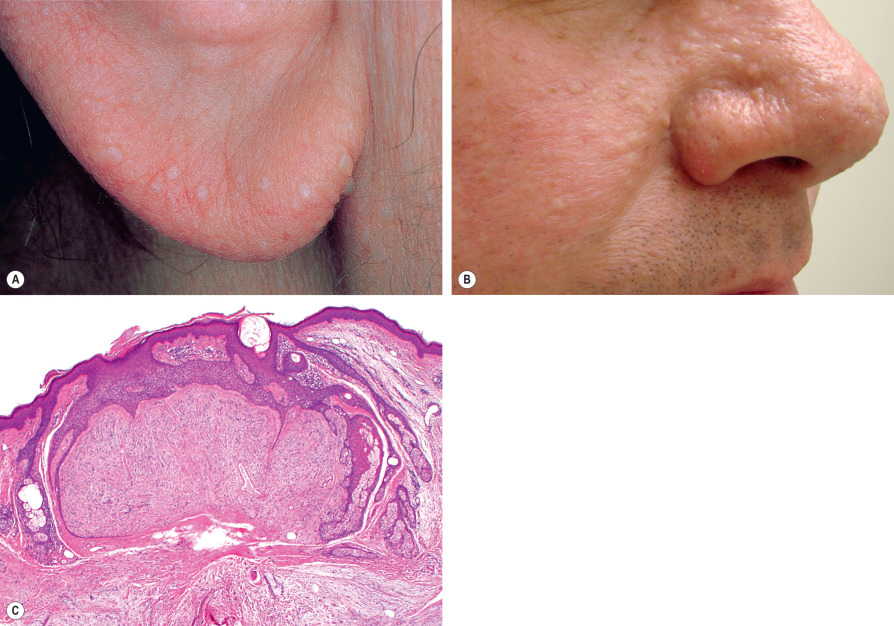
| EVALUATION OF PATIENTS WITH BIRT–HOGG–DUBÉ SYNDROME (BHDS) |
|
Multiple papules of the face and pinnae can also be seen in patients with familial multiple discoid fibromas, but there are no systemic manifestations or mutations in FLCN . Mutations have been detected in FNIP1 , when encodes a folliculin interacting protein.
Pathology
At low magnification, fibrofolliculoma displays slender strands of follicular mantle cells that emanate from a folliculosebaceous unit at the level of the isthmus ( Fig. 111.4C ). Usually, the strands are composed of cells with a slightly basaloid appearance, and sometimes, tiny integrated collections of mature sebocytes are identifiable. The epithelial component may assume a mitt-like morphology in some instances, encasing the vascularized fibrous stroma.
Perifollicular fibroma consists almost exclusively of stroma that is identical to the stromal elements of fibrofolliculoma and angiofibroma. Lesions consist of spindled and stellate cells arrayed concentrically around follicles and distributed amongst thickened collagen bundles with a proportionate number of intervening small thin-walled vessels. Sometimes, a concurrent component of follicular epithelial hyperplasia is identifiable, although the epithelium tends to be inconspicuous in comparison to fibrofolliculoma. Trichodiscoma was thought by Pinkus to represent a unique proliferation with differentiation toward the haarscheibe (hair disk), which is probably a mythical structure. More recently, trichodiscomas have been interpreted as fibrofolliculomas or perifollicular fibromas with myxoid rather than fibrous stroma. Lastly, the so-called neurofollicular hamartoma is now considered to be a type of trichodiscoma.
Treatment
Fibrofolliculomas, perifollicular fibromas, and trichodiscomas are all benign and require no surgical treatment. Superficial electrodesiccation, laser ablation, or dermabrasion can be utilized to ameliorate multiple lesions. Unlike the angiofibromas of tuberous sclerosis, topical rapamycin did not result in cosmetic improvement in the fibrofolliculomas of Birt–Hogg–Dubé syndrome .
Nevus sebaceus
Introduction
Originally known as organoid nevus , nevus sebaceus is commonly thought of as a sebaceous lesion and historically has been the first entity discussed in chapters describing sebaceous tumors. In truth, nevus sebaceus is a non-neoplastic malformation that includes follicular, sebaceous and apocrine elements as well as epidermal hyperplasia.
Clinical features
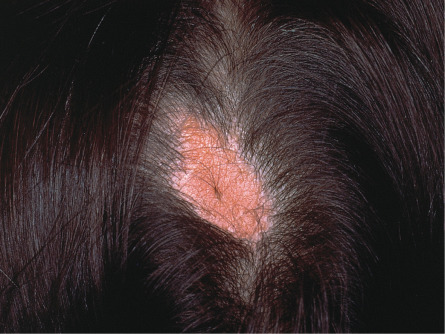
Nevus sebaceus represents a classic nevus or congenital malformation. At birth, lesions are only slightly raised and subtly discernible. Nevus sebaceus involves the scalp or face commonly, the neck occasionally, and the trunk rarely. Lesions that are linear are distributed along the lines of Blaschko, although this may be difficult to appreciate if small in size. On the scalp, a nevus will remain hairless, or mostly so, as the infant’s hair grows normally around it. During childhood, the nevus thickens slightly and assumes a yellow or orange hue ( Fig. 111.6 ). At adolescence, progressive thickening occurs and the surface becomes pebbly or verrucous. Especially when there is more extensive involvement with nevus sebaceus, the possibility of Schimmelpenning syndrome or phakomatosis pigmentokeratotica needs to be considered (see Table 62.7 ).
In a study of 65 sebaceous nevi, postzygotic somatic mutations were detected in HRAS (95% of nevi) and KRAS (5%) . Mosaicism for HRAS and KRAS mutations was also detected in individuals with Schimmelpenning syndrome. The more common mutations were shown to lead to activation of the MAPK and PI3K-Akt pathways (see Ch. 113 ). A syringocystadenoma papilliferum or trichoblastoma that arises within a nevus sebaceus carries the same HRAS mutation as the underlying nevus .
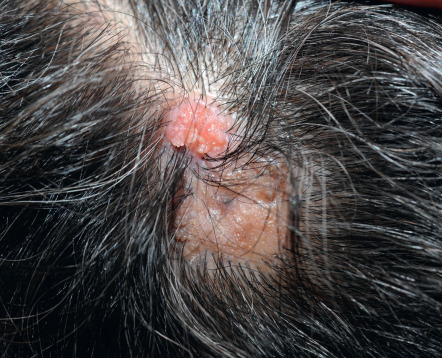
Nevus sebaceus represents a fertile field for the development of secondary adnexal neoplasms, commonly benign but occasionally malignant . Past generations of dermatologists were taught that secondary carcinoma develops in 10% or more of these lesions over time, although this figure was poorly substantiated. The vast majority of secondary proliferations represent benign follicular germinative (trichoblastoma-like) foci; the actual incidence of secondary basal cell carcinoma (BCC) is <1% . Historically, syringocystadenoma papilliferum (papillary syringoadenoma) was believed to represent the most common secondary proliferation ( Fig. 111.7 ), but recent analyses indicate that trichoblastoma occurs more frequently . Other common secondary neoplasms include tricholemmoma, desmoplastic tricholemmoma, sebaceous adenoma, apocrine adenoma, and poroma. Only rarely does secondary sebaceous carcinoma or apocrine carcinoma arise in a nevus sebaceus. Malignancy probably develops only in longstanding or neglected lesions but exceptionally can be a source of mortality.
When a nodule develops within a nevus sebaceus and large blue–gray ovoid nests are noted by dermoscopy, then the pattern of the nests should be assessed. If the entire lesion is a blue–gray nest (symmetric pattern) trichoblastoma is favored whereas an asymmetric pattern of nests favors BCC.
Pathology
In nevus sebaceus, the principal malformation is of individual folliculosebaceous units. An infantile nevus sebaceus can be microscopically subtle, particularly if the biopsy specimen includes only lesional skin, as the malformed follicular units are small and deviate little from normal. In an excisional specimen from the scalp during later childhood, tiny misshapen lesional follicles provide a stark contrast to normal terminal follicles at the periphery of the biopsy. Small apocrine glands may be identifiable in a specimen of sufficient depth. The epidermis also thickens and becomes progressively more papillated, gradually assuming a configuration akin to an epidermal nevus.
By late adolescence, nevus sebaceus displays an epidermal pattern identical to that of an epidermal nevus, typified by acanthosis and fibroplasia of the papillary dermis. There may be enlarged sebaceous glands in contiguity with the dermal–epidermal junction ( Fig. 111.8 ). Buds of follicular germinative cells may jut from the junction as well. The underlying follicular units remain smallish and appear distorted but their sebaceous lobules increase in prominence. Dilated apocrine glandular structures with inspissated secretions are not uncommonly identifiable in the reticular dermis. This histopathology remains stable into adulthood.
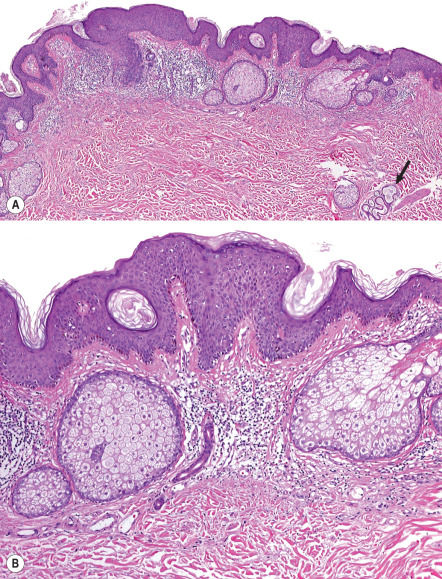
Over time, some patients develop exaggerated verrucous epidermal hyperplasia much like a verruca vulgaris. It has been suggested that this is a consequence of human papillomavirus infection . Secondary neoplasms are common, and trichoblastoma, tricholemmoma, and syringocystadenoma are frequently observed . The strong association between syringocystadenoma and nevus sebaceus warrants careful scrutiny any time the diagnosis of syringocystadenoma is rendered, to ascertain whether a nevus sebaceus can be found in the same specimen.
Treatment
As carcinoma was formerly reputed to commonly develop within nevus sebaceus, the risk of transformation was used as justification for complete excision. In truth, the risk of development of secondary carcinoma is low . However, the risk for the development of a secondary benign neoplasm, such as syringocystadenoma or trichoblastoma, is relatively high. In addition, it is clear that nevus sebaceus becomes increasingly verrucous and conspicuous over time. In light of this, conservative complete excision during late childhood represents a commonly utilized, but not required, approach. Lesions within the scalp may be difficult for the patient to follow clinically. For facial lesions, consideration can be given to excision during childhood, before the development of secondary verrucous alteration. The excisional margins can be narrow. Removal by shave or laser ablation is usually not successful.
Mixed tumor (chondroid syringoma)
Introduction
Cutaneous mixed tumor, also known as chondroid syringoma , represents an acquired hamartoma with folliculosebaceous-apocrine differentiation that has been interpreted as an adnexal adenoma since its description. Despite this legacy, in the opinion of the authors, mixed tumor is best pigeonholed as a hamartoma. The stroma of a mixed tumor is always abundant and generally comprises half of the surface area of a given lesion in conventional sections, and the epithelial component is commonly heterogeneous . An analogy has been drawn between cutaneous mixed tumor and mixed tumor (pleomorphic adenoma) of the salivary gland. While the comparison has some validity, it is important to note that pleomorphic adenoma of the salivary gland functions as a neoplasm in a biological sense, as it holds a tendency for local recurrence if incompletely extirpated as well as the potential for evolution to malignancy. In contrast, chondroid syringoma represents an indolent process virtually devoid of proliferative capacity, and it rarely recurs after enucleation.
Some authors consider chondroid syringoma and cutaneous myoepithelioma to represent two ends of a spectrum, and this concept is supported by the fact that some chondroid syringomata are particularly rich in myoepithelial cells . In addition, some authors consider cutaneous and salivary mixed tumors to be related myoepithelial neoplasms because both can have gene rearrangements involving PLAG1 (pleomorphic adenoma gene 1), which leads to overexpression of PLAG1, a zinc finger transcription factor .
Clinical features
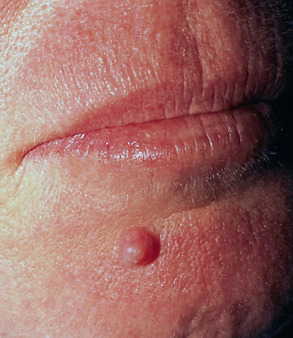
The clinical presentation of a mixed tumor is indistinctive ( Fig. 111.9 ). The lesions present as cutaneous papulonodules that are often misconstrued as cysts. Involvement of the head and neck is common and lesions may also develop on the trunk or within axillary or genital skin.
Pathology
Mixed tumor presents pathologically as a well-circumscribed nodule that resides within the deep reticular dermis or subcutis. Its biphasic morphology includes epithelial structures encompassed by ample varied stroma . There is always a prominent collagenous component to the stroma, but myxoid and lipocytic zones can also be found. Despite its name, stromal hyaline cartilage is found in less than half of all chondroid syringomata.
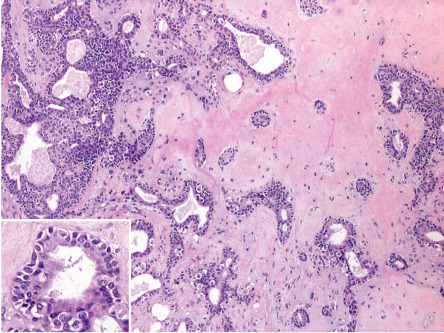
The epithelial component of a mixed tumor displays two primary patterns. Most mixed tumors demonstrate large inter-anastomosing and branching tubules lined by two cell layers – columnar cells with decapitation secretion at the luminal border and cuboidal cells peripherally; this configuration has been dubbed apocrine mixed tumor ( Fig. 111.10 ). Less commonly, mixed tumors show an epithelial component that includes many small, rounded, cuticulated ducts ( Fig. 111.11 ). Such lesions have been interpreted by some as eccrine. Rather, it seems likely that all mixed tumors are hamartomas of folliculosebaceous-apocrine lineage and that the configuration of the epithelial component can vary.
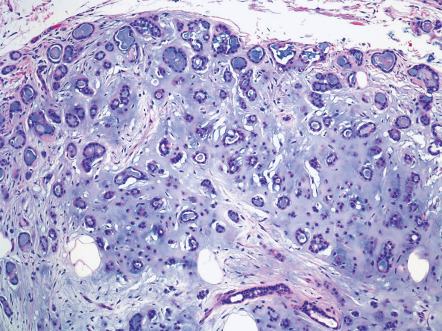
The epithelial component of an apocrine mixed tumor also commonly includes follicular and sebaceous elements . Follicular differentiation can consist of small clusters of follicular germinative cells, lobules of outer sheath cells, cystic foci with isthmic or infundibular keratinization, and clusters of matrical cells. Small collections of sebocytes may also be found.
The number of myoepithelial cells in chondroid syringomas varies. Rare tumors composed exclusively of myoepithelial cells are classified as cutaneous myoepithelioma (see below).
Treatment
Mixed tumor is benign and indolent. Most lesions are treated by simple enucleation, and the risk for persistence/recurrence is minimal. In contrast to mixed tumors of salivary origin, there is no need for excision with margins.
Neoplasms and Proliferations With Follicular Germinative Differentiation
Trichoepithelioma/trichoblastoma
Introduction
Trichoepithelioma and trichoblastoma are terms that refer to benign neoplasms with mostly follicular germinative differentiation . Historically, the term trichoepithelioma is older and represents the name ascribed to a solitary lesion of the multiple familial form of trichoepithelioma (epithelioma adenoides cysticum), and thus the term trichoepithelioma is clinically useful in this context. The designation trichoblastoma was coined more recently and was originally applied to neoplasms that showed follicular bulbar differentiation almost exclusively. Although trichoepithelioma is still used, trichoblastoma has evolved to be an overarching label for benign proliferations with follicular germinative differentiation . For many, in the current dermatopathologic lexicon, trichoepithelioma represents a variant of trichoblastoma.
Various authors have described subtle variations within the spectrum of follicular germinative neoplasms, thereby birthing terms such as trichogerminoma, trichoblastic fibroma, and lymphadenoma (adamantinoid trichoblastoma) . For purposes of this discussion, these variants will be considered as falling within the larger spectrum of trichoblastoma.
Clinical features
Classic trichoepithelioma usually presents as a skin-colored papule or nodule on the face or upper trunk. Lesions have a predilection for the nose ( Fig. 111.12 ). When in multiplicity, constituting epithelioma adenoides cysticum or Brooke disease, lesional density is always greatest in the central face. The cylindromatosis gene ( CYLD ), whose protein product functions as a tumor suppressor, underlies Brooke disease. CYLD encodes a deubiquitinating enzyme that impedes the nuclear factor (NF)-κB and c-Jun N-terminal kinase (JNK) pathways . Specifically, missense mutations lead to amino acid substitutions within the ubiquitin (Ub)-specific protease (USP) domain of the CYLD protein. Brooke disease can be cosmetically troublesome and is associated with a risk of secondary BCC . Patients can also develop cylindromas and spiradenomas (see Fig. 111.5 ). Lesions described historically as trichoblastoma were often larger and deeper than simple trichoepithelioma.
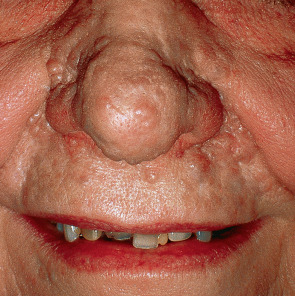
Pathology
All trichoepitheliomas and trichoblastomas have in common the architectural attributes of a benign neoplasm – namely, relative symmetry, circumscription, and a lack of substantial cytologic atypicality. In addition, all are linked by prominence of follicular germinative cells with enveloping fibrocytic stroma that varies in degree . In classic trichoepithelioma, the fibrocytic stroma is conspicuous and constitutes as much as half of the cellularity of the lesion in a given cross-section. In other lesions that simulate BCC, follicular germinative cells may be arrayed as either small or large nodules with only scant intervening sclerotic stroma. The coarse fibrous stroma typically maintains tightly adherent contact to the lesional follicular germinative cells . This is in distinct contrast to the pattern of BCC, in which clefts between basaloid cells and stromal elements often serve as a diagnostic clue.
In classic trichoepithelioma, follicular germinative cells are often disposed as small clusters or as reticulate and cribriform cords ( Fig. 111.13 ). There are usually foci of pronounced bulbar differentiation, emulating the follicular bulb and papilla; these structures have been termed papillary mesenchymal bodies . Classic trichoepithelioma does not typically show follicular germinative differentiation exclusively. Rather, small cornifying cystic spaces with surrounding pinkish keratinocytes, reflecting concurrent infundibular or isthmic differentiation, are also apparent.
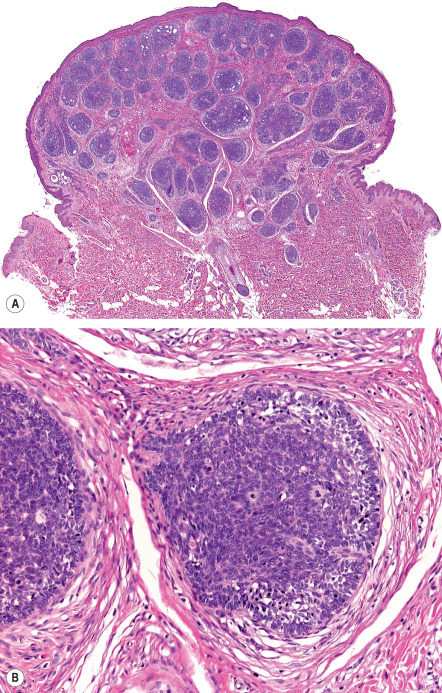
In small or large nodular trichoblastoma, follicular germinative cells comprise the majority of the surface area, papillary mesenchymal bodies may be inconspicuous, and superficial follicular differentiation may be absent. Trichoblastic fibroma is a designation used to characterize small nodular trichoblastomas with exaggerated fibrous stroma that often constitutes over half of the cross-sectional area of the lesion . Papillary mesenchymal bodies may be conspicuous in trichoblastic fibroma. With close scrutiny, other modes of differentiation can occasionally be found in a trichoblastoma. There may be tiny collections of mature sebocytes, reflecting concurrent sebaceous differentiation, and (apocrine) ductal differentiation may also be noted.
The differential diagnosis often includes BCC and differentiation is best accomplished by light microscopy. Nonetheless, the role of immunohistochemical staining with markers such as CK20 and PHLDA1 ( p leckstrin h omology- l ike d omain, family A , member 1 ) protein, a follicular stem cell marker, has been investigated. Positive CK20 labeling of Merkel cells colonizing basaloid aggregates has been shown to favor trichoepithelioma/trichoblastoma over BCC , and positive staining for PHLDA1 protein was observed in desmoplastic trichoepitheliomas but not in morpheaform BCCs . However, a subsequent study found that while morpheaform BCCs did not express PHLDA1, a sizable minority of micronodular BCCs did , thus this marker appears to be most helpful if the pattern in consideration is a morpheaform BCC.
Treatment
Trichoblastoma is benign and provokes no imperative for surgical treatment. Multiple facial trichoepitheliomas can be cosmetically disabling and many affected patients desire some type of intervention. Longitudinal evaluation for the possibility of secondary BCC is warranted . Because of the number of lesions, conventional excision is not indicated. Other ablative approaches, including laser or electrosurgical destruction, have been employed with some success .
Desmoplastic trichoblastoma (desmoplastic trichoepithelioma)
Introduction
Originally termed sclerosing epithelial hamartoma , desmoplastic trichoepithelioma represents a variant of trichoblastoma with exaggerated stromal sclerosis (desmoplasia) . It can easily be misinterpreted as morpheaform BCC by the uninitiated.
Clinical features
Desmoplastic trichoepithelioma commonly presents as a firm skin-colored to erythematous annular plaque with a central dell. The cheek of a woman represents a stereotypical context . Most examples do not exceed 1 cm in diameter. Nearly all desmoplastic trichoepitheliomas are solitary. Multiplicity is a rarity.
Pathology
Desmoplastic trichoepitheliomas are composed of cords of basaloid cells, often two cells wide, arrayed interstitially amongst coarsened collagen bundles ( Fig. 111.14 ) . The lesions are relatively circumscribed and are often confined to the upper two-thirds of the reticular dermis, although this is difficult to appreciate in biopsies en parte . Small cystic foci of isthmic or infundibular keratinization may be present, and if these cystic spaces are predominant, the designation trichoadenoma is apt. The cornifying cysts can rupture and elicit a granulomatous reaction, and small secondary foci of calcification may also be identified .
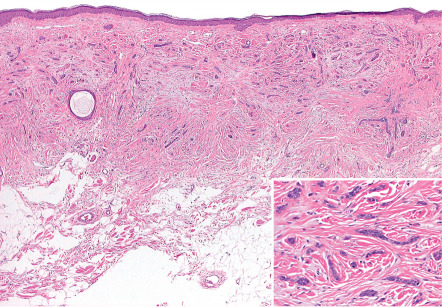
Treatment
Desmoplastic trichoepithelioma is benign. Small or partial biopsies may yield diagnostic uncertainty with respect to morpheaform BCC or microcystic adnexal carcinoma as a consideration in the differential diagnosis, and re-excision or resampling may be necessary when the interpretation is ambiguous. Negative staining for PHLDA1 (see previous section) supports the diagnosis of morpheaform BCC.
Neoplasms and Proliferations With Matrical Differentiation
Pilomatricoma
Introduction
Pilomatricoma (pilomatrixoma, trichomatrioma, calcifying epithelioma) is a benign neoplasm or cyst typified by follicular matrical cornification. It has been demonstrated that mutations in CTNNB1 , the gene that encodes β-catenin, a component of a key signaling pathway that influences cell differentiation and proliferation, are generally present in matrical neoplasms, including pilomatricomas . Pilomatricomas also commonly demonstrate trisomy 18 .
Clinical features
Pilomatricoma usually presents as a solitary skin-colored or bluish nodule ( Fig. 111.15 ). Rarely, multiple lesions are evident, sometimes in association with myotonic dystrophy, Turner syndrome, or Gardner syndrome (when cystic). Firmness is typical and reflects secondary calcification as well as accompanying fibrosis and granulomatous inflammation. Some lesions are “rock-hard” and plate-like on palpation. An angulated shape can be appreciated by stretching the overlying skin (“tent sign”). Although pilomatricomas may develop on any hair-bearing surface, the majority occur on the head or upper trunk . Most tumors appear during childhood and adolescence, but pilomatricomas may develop at any age and can simulate BCC, clinically or microscopically, especially in later adulthood. Occasionally, the dermis overlying a pilomatricoma will develop anetoderma.
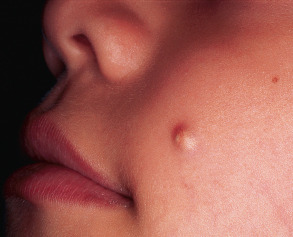
Pathology
Histopathologically, an early pilomatricoma frequently presents as a cyst with central matrical cornification. The cyst wall consists of basaloid matrical cells that show an abrupt transition to central eosinophilic, cornified matrical cells in which barely discernible nuclear outlines remain ( Fig. 111.16 ). Sometimes, pink trichohyalin granules, illustrative of matrical cornification, are identifiable at the transition point. The central anucleate cornified cells are commonly referred to as ghost or shadow cells.
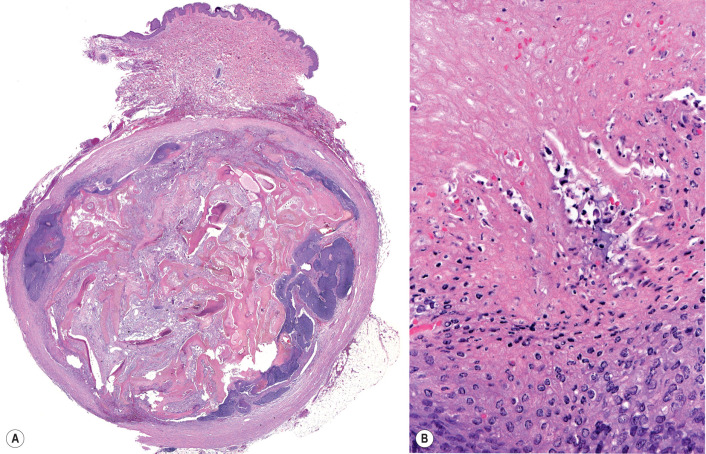
In fully developed pilomatricomas, a cystic configuration is commonly lost. Solid collections of basaloid matrical cells and matrical corneocytes are present in varying degrees. The cornified matrical cells elicit considerable fibrosis and a secondary granulomatous infiltrate, which can become predominant in longstanding lesions. Matrical cells have a proliferative capacity as high as any human tissue and can display numerous cells in mitosis. The designation proliferating pilomatricoma refers to a pilomatricoma with a high mitotic index. At times, such lesions can be misinterpreted as carcinoma . In late lesions, basaloid matrical cells may be lacking. An involutional pilomatricoma at times can present with only a few shadow cells buried in a larger fibrosing granulomatous reaction. Calcification and ossification also ensue in late lesions.
Treatment
Pilomatricoma is a benign lesion that is usually treated by simple enucleation. A pilomatricoma may recur after limited excision. If multiple recurrences are observed, complete excision (with negative margins) should be performed to exclude pilomatrical carcinoma.
Pilomatrical carcinoma
Introduction
Pilomatrical (matrical) carcinoma is an uncommon malignancy that exhibits matrical cornification, much like pilomatricoma . Mutations in CTNNB1 (encodes β-catenin) are common in matrical carcinoma . This suggests a partial common pathogenesis for pilomatricoma and matrical carcinoma. Although on a practical basis no clear association exists, there is a theoretical risk for the possibility of matrical carcinoma developing from conventional pilomatricoma.
Clinical features
Matrical carcinoma is typically a disease of adulthood. Lesions develop on the head and neck, often in the retroauricular area. Although a pathogenic link with ultraviolet irradiation has not been established, many matrical carcinomas develop within severely sun-damaged skin. If a diagnosis of matrical carcinoma is offered in a child or young adult at an age when the diagnosis is improbable, the case should be reassessed for the possibility of pilomatricoma.
Pathology
Much like pilomatricoma, matrical carcinomas include both basaloid matrical cells and anucleate matrical corneocytes. In contrast to pilomatricoma, the matrical carcinoma cells usually display nuclear pleomorphism. An infiltrative configuration or poor circumscription may be apparent, especially in an excisional biopsy. There may be considerable squamous cornification as well, and parts of a given lesion may resemble conventional squamous cell carcinoma (SCC).
Matrical carcinoma should be distinguished microscopically from BCC with matrical differentiation . Histopathologically, the latter entity represents an otherwise conventional BCC in which small foci of matrical cornification are identifiable. The clinical behavior of this variant is thought to be identical to that of conventional BCC.
Treatment
Matrical carcinoma is a low-grade form of adnexal carcinoma that poses some risk for metastasis, although the risk is believed to be low . Complete extirpation with negative margins represents standard care.
Neoplasms and Proliferations With Follicular Sheath (Tricholemmal) Differentiation
Tricholemmoma
Introduction
Tricholemmoma (trichilemmoma) constitutes a form of benign adnexal neoplasm with differentiation toward the follicular outer sheath . Some have averred that tricholemmoma is merely a viral wart based upon conventional histopathology . Generally, molecular analysis has failed to demonstrate intralesional papillomavirus DNA and thus viral causation seems unlikely .
Clinical features
Tricholemmomas may arise as simple or as multiple papules that usually have similar coloration to surrounding normal skin (see Fig. 63.3 ) . Individual lesions may display hyperkeratosis or a verrucous surface. Most are distributed on the central face, including the nose or upper lip, but any non-glabrous site may be affected. Tricholemmomas can also present on genital skin and simulate condylomata acuminata if multiple. Tricholemmoma may occur as a secondary neoplasm within nevus sebaceus. The variant desmoplastic tricholemmoma is especially common in this context .
Multiple tricholemmomas typically present on facial or genital skin. Multiple tricholemmomas are a common manifestation of Cowden syndrome (see Ch. 63 & Fig. 111.5 ) . In addition to tricholemmomas, manifestations of Cowden syndrome include sclerotic fibromas, acral keratoses, and adenocarcinoma of the breast, thyroid gland or gastrointestinal tract . Loss of PTEN expression within a tricholemmoma, as assessed by immunohistochemistry, is suggestive that the patient has Cowden syndrome .
Pathology
Microscopically, tricholemmomas present as small circumscribed lobular proliferations composed of pallid, glycogen-containing outer sheath cells. There is often a broad connection to the surface epidermis ( Fig. 111.17 ) . A papillated surface is common, and there may be verrucous hyperplasia with hypergranulosis, thereby simulating the pattern of a verruca in a superficial biopsy. Squamous eddies are not uncommon. There is a palisade of small, compact basal keratinocytes at the periphery of a lesional lobule, and a flanking thickened basement membrane that is PAS-D (periodic acid Schiff with diastase)-positive is commonly present. Outer sheath differentiation can also be recognized through use of CD34 immunostaining.