24 Scalp, Cheek, and Neck Deformities
Summary
Pediatric scalp, cheek, and neck deformities include: cutis aplasia, alopecia, hemifacial atrophy, and webbed neck. Skin lesions and trauma also may require reconstruction of these areas. Intervention is based on the type of deformity and the age of the patient.
24.1 Introduction
Reconstruction of pediatric scalp, cheek, and neck deformities can be performed by several methods. Scalp defects may be congenital (e.g., cutis aplasia), but more commonly result from extirpation of lesions or trauma. The most frequent congenital malformation of the cheek is hemifacial microsomia (covered in another chapter), followed by progressive hemifacial atrophy. Neck lesions such as branchial cleft anomalies and thyroglossal duct cysts usually are managed by pediatric general surgeons or otolaryngologists. Plastic surgeons are asked to improve soft-tissue neck deformities, such as webbing associated with Turner syndrome.
24.2 Diagnosis
Diagnosis of scalp, cheek, and neck deformities is made by history and physical examination. Imaging rarely is necessary, but may be indicated for cutis aplasia or hemifacial atrophy to determine if an underlying osseous abnormality exists. Occasionally, histopathology aids the diagnosis of scalp lesions.
24.3 Nonoperative Treatment
The primary morbidity of scalp, cheek, and neck deformities is psychosocial. Although cutis aplasia may require immediate operative intervention, other disorders can be observed. Timing of intervention typically involves three periods: (1) infancy, (2) between 3 and 4 years of age, and (3) late childhood or adolescence. Large lesions of the scalp are best removed during infancy because scalp redundancy at this time facilitates extirpation and reconstruction. If a deformity is likely to cause decreased self-esteem, then improving the condition between 3 and 4 years of age should be considered. Because long-term memory and self-esteem begin to form at approximately 4 years of age, correcting a deformity at age 3 often is desired. Some parents prefer to wait until children are old enough to decide whether or not they would like to have an operation, especially if the deformity is minor. Patients typically do not request a procedure until late childhood or early adolescence because before this time the fear of an operation outweighs their desire to improve a deformity.
24.4 Operative Treatment
24.4.1 Scalp
Cutis Aplasia
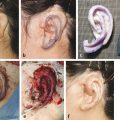
Cutis aplasia is a rare condition (1/5,000 newborns), and includes absence of scalp soft tissues, bone, and/or dura. (Fig. 24‑1). The disorder can affect any area of the body, but the scalp is the most common site (84%). The defect usually is along the sagittal suture and the area is covered with a thin membrane. One-fourth of patients have an underlying osseous or dural defect and thus are at risk for infection, venous thrombosis, and/or hemorrhage. Because cutis aplasia may be associated with syndromes and other anomalies, patients should be evaluated for additional medical conditions.
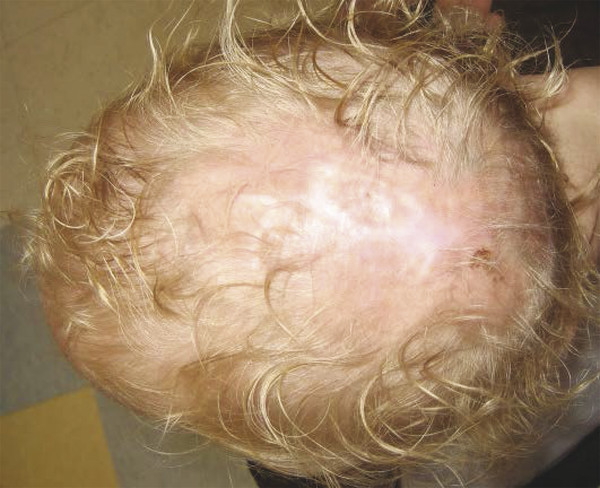
Management of cutis aplasia is based on the depth of the defect. If the bone is intact, superficial tissues are treated with local wound care and the area heals secondarily. Alopecia can be improved at a later date. If a cranial defect is present with exposed dura, the condition is life-threatening. Operative intervention is necessary within the first few hours after birth to prevent desiccation of the dura, meningitis, thrombosis of the sagittal sinus, and/or hemorrhage from exposed veins. If possible, the area should be closed by wide subgaleal undermining of scalp flaps. However, most defects are too large to close linearly and require a split-thickness skin graft over the dura. The thin graft is harvested from the gluteal area and is meshed. It is important to graft the dura before an eschar has formed. Once an eschar has developed, it is difficult to remove and the child is at risk for life-threatening hemorrhage and dural injury from debridement. If an eschar is present, it is best elevated off the dura using hydrodissection. Rarely, cutis aplasia can result in exposed brain without dura; the neonate requires emergent flap coverage of the area.
Because osseous defects spontaneously heal in infancy, cranioplasty is not performed. Most bone gaps will close and rarely require secondary reconstruction. If a large osseous defect remains, it can be filled with particulate bone graft any time after 18 months of age when the dura no longer has the ability to create new bone.
Following closure of the soft-tissue defect, alopecia can be managed electively. Because the infant scalp has significant redundancy during infancy as the brain is rapidly enlarging, serial excision of the scar or graft should be considered starting at 6 months of age. If the area is not amenable to serial excision, then tissue expansion can be performed later in childhood. Small areas of alopecia can be observed until the child is older to determine if it will cause psychosocial morbidity.
24.4.2 Skin Lesions
Because most brain growth occurs during the first year of life, the scalp has unique redundancy during this period. Consequently, large skin lesions are best removed during infancy to facilitate the procedure and to achieve the most favorable outcome. I begin excision at 6 months of age when the risk of anesthesia is equivalent to an adult. If the lesion is very large and will take multiple serial resections, I perform the first resection at 3 months of age (Fig. 24‑2). The operations are performed at 6-week intervals when the wound has achieved 80% of its strength, and the adjacent skin has had sufficient time to relax. In some cases, the incision line cannot be completely closed after widely undermining skin flaps and performing galeal scoring. Small openings (e.g., 1–2 cm) can be left open to heal secondarily.

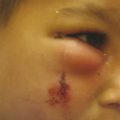
Most scalp lesions can be removed by serial excision. However, some wounds require skin graft reconstruction if (1) the lesion is large, (2) the child has minimal scalp laxity, (3) the defect is in an area where it is difficult to advance skin flaps from two directions (e.g., ear), and (4) the lesion is unable to be partially excised because suturing remaining diseased tissue together under tension is problematic (e.g., neurofibroma, vascular malformation; Fig. 24‑3). A split-thickness skin graft contracts, giving a wound that is approximately one-third smaller than the original defect. The skin graft then can be removed later by serial excision or tissue expansion.

Another option to reconstruct large defects following lesion removal is to expand the scalp prior to excision. This is a reasonable approach for nevi, but should not be performed for vascular lesions (e.g., neurofibroma, vascular anomalies) because tissue expansion can stimulate the disorder by increasing neovascularization; I prefer to remove the lesion, obtain a smaller abnormality with a skin graft, and then reconstruct the area secondarily with tissue expansion. I attempt to avoid tissue expansion whenever possible because it is traumatic to patients/families and is associated with significant complications (e.g., infection, extrusion). However, tissue expansion is the only alternative to reconstruct hair-bearing scalp for large areas not amenable to serial excision.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


