1 Craniosynostosis
Summary
Craniosynostosis is the premature fusion of one or more cranial sutures. It can occur in isolation (non-syndromic) or in association with a syndrome (syndromic), and can involve a single or multiple cranial sutures. Patients with non-syndromic single suture fusions have normal intelligence, but may display subtle learning disabilities. Patients with a syndromic diagnosis and/or multiple suture fusions tend to have variable degrees of cognitive impairment. The sagittal suture is the most commonly (40 to 55%) fused suture followed by the coronal (20 to 25%), metopic (5 to 15%), and lambdoid (1 to 5%) sutures. This condition has two primary consequences: a characteristic change in cranial shape and, more importantly, potential restriction of cerebral growth and development. The morphologic effects of suture fusion are related to the location and number of fused sutures, and the age at which the fusion(s) occurred. Diagnosis is typically made by physical examination (i.e., the shape of the head) and/or computed tomography. The general goals of treatment are to normalize cranial shape and intracranial volume, but other considerations (e.g., respiration, ophthalmologic, facial aesthetics, musculoskeletal) may require additional intervention. Treatment options to address the craniosynostosis include: minimally-invasive procedures (endoscopically-assisted strip craniectomy (SC), SC+ spring mediated distraction), open craniotomy procedures (Pi, open release), cranial distraction, and open cranial remodeling with osseous stabilization. Results of treatment vary according to the presence/absence of an associated syndrome, age at procedure, type of procedure used, duration of follow-up.
1.1 Introduction
Craniosynostosis is defined as the premature fusion of one or more cranial sutures. This condition occurs in an estimated 1 in 2,000 to 2,500 live births, and can occur as a solitary finding (termed nonsyndromic craniosynostosis) or in conjunction with pathologically associated physical anomalies (syndromic). The majority (80–95%) of affected patients have a single fused suture and no concurrent syndrome. Of the patients with multiple suture fusions, approximately 75% have an associated syndrome. The sagittal suture is the most commonly involved (40–55%), followed by the coronal (20–25%), metopic (5–15%), and lambdoid (1–5%) sutures. Some recent reports document a rise in the incidence of isolated metopic fusion and place it second behind sagittal synostosis; this finding is not universally reported and may reflect diagnostic variability between providers and centers and not a true rise in pathoanatomy. Fusions of other sutures (e.g., squamosal and frontosphenoidal) are much less common.
Craniosynostosis has two primary consequences: a characteristic change in cranial shape and, more importantly, potential restriction of cerebral growth and development. The morphologic effects of suture fusion are related to the location and number of fused sutures and the age at which the fusion(s) occurred. Most documented forms of craniosynostosis occur prenatally, and malformation of the craniofacial skeleton is evident at birth. However, there are rare examples of abnormal sutural fusion that occur after birth; in such instances, the changes in cranial shape may be subtle and difficult to diagnose.
1.2 Cranial Development, Anatomy, and Pathoanatomy
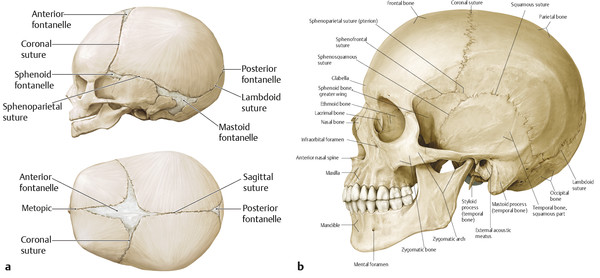
Cranial sutures and fontanelles are the mesenchymes that persist between the calvarial plates. The major cranial sutures are the metopic, sagittal, coronal, and lambdoid sutures (Fig. 1‑1). Minor cranial sutures include the temporosquamosal, frontonasal, sphenoethmoidal, and the frontosphenoidal sutures. The cranial sutures have two primary functions: to permit cranial deformation or molding during parturition so that the infant can pass through the pelvis and to allow the cranium to accommodate accelerated brain development early in life. As the brain grows, tension across the suture increases and stimulates bone deposition along the edges of the calvarial plates. Thus, cranial growth occurs in response to brain growth. Once brain growth is completed, the cranial sutures serve little purpose and eventually close. Conventional wisdom suggests that the cranial sutures close in childhood, but histologic sections suggest that true bony fusion occurs much later.
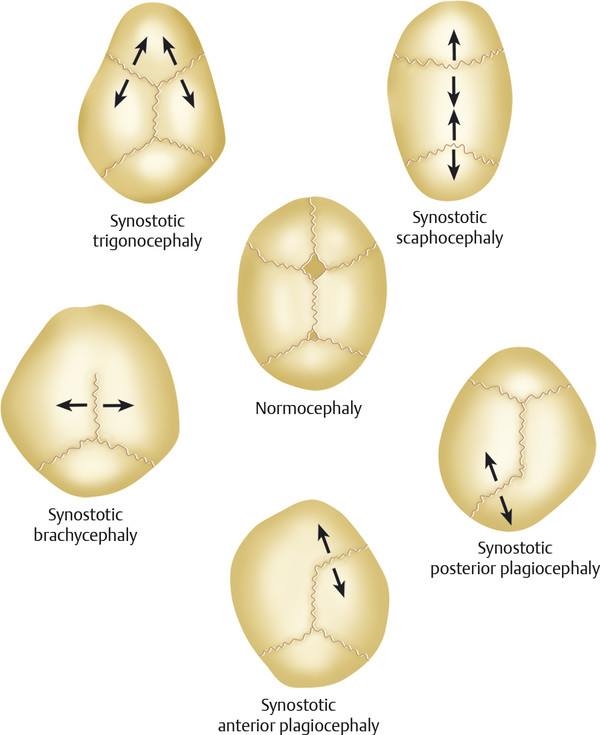
The complex biological activity of the cranial sutures is still being elucidated. Sommering (1800) was the first to describe the anatomy of the cranial sutures and to propose that premature fusion of the suture could alter cranial shape. Rudolph Virchow (1851) expanded his work and posited that cranial suture fusion (or patency) was independent of the perisutural environment. He further hypothesized that premature suture fusion resulted in compensatory skull growth parallel to the fused suture and a decrease in growth perpendicular to the suture (Virchow’s law; Fig. 1‑2). Although Virchow’s law is the foundation for clinical diagnosis of craniosynostosis, the understanding of sutural biology has changed dramatically. Beginning with the work of various researchers, including Park and Powers (1920), Van der Klaauw (1946), and Moss (1959), attention began to focus on the influence of the dura mater on suture activity, patency, and closure. Numerous in vitro and in vivo models strongly suggest that the underlying dura mater medicates suture activity through temporal and spatial production of growth factors (e.g., fibroblast growth factor [FGF]-2) and cellular elements (e.g., osteoblastic cells) to the overlying osteogenic fronts and suture mesenchyme.
Genetic methods have now identified more than 100 mutations that are involved in the pathogenesis of craniosynostosis, including TWIST, NELL-1, MSX-2, GLI3, and fibroblast growth factor receptor (FGFR)-1 to -3. These are covered more extensively in Chapter 2 on craniofacial syndromes. The mechanism through which these mutations cause craniosynostosis is still being elucidated. Craniosynostosis appears to affect facial growth through a secondary cascade of growth impairment that extends from the cranial base into the facial skeleton. Findings by Mooney and others suggest that the calvarial dysmorphology can drive the basicranial and midface changes. The controlling influence of genetics cannot be minimized, but epigenetic influences and mechanotransduction can modulate genetic expression and patient phenotype. There are well-documented examples of individuals who possess genes associated with certain syndromic forms of craniosynostosis (e.g., FGFR-3 Pro250Arg and TWIST) and yet lack any evidence of cranial fusion or the phenotypic features of the underlying syndrome.
1.3 Classification
Craniosynostosis can be generally classified based on both the number of involved sutures (single vs. multiple) and the presence or absence of an associated syndrome (nonsyndromic vs. syndromic). In general, most patients (~85%) with single-suture craniosynostosis have no identifiable associated syndrome (nonsyndromic, single-suture craniosynostosis), whereas most patients (~75%) who carry a syndromic diagnosis have fusion of two or more sutures (syndromic, multisuture craniosynostosis). However, it is incorrect to use the terms single-suture and nonsyndromic or multisuture and syndromic interchangeably. There are many examples of patients with isolated sutural fusions who have an associated syndromic diagnosis (syndromic, single-suture craniosynostosis) and patients with multisutural fusion that have no identifiable syndrome (nonsyndromic, multisuture craniosynostosis). Approximately 25% of patients with unilateral coronal synostosis have an associated molecular/syndromic association: Muenke’s syndrome (FGFR-3 Pro250Arg; most common), Apert’s syndrome (FGFR-2), Saethre–Chotzen syndrome (TWIST) and craniofrontonasal syndrome (EFNB1). Similarly, an estimated 28% of patients with metopic craniosynostosis have an associated syndrome or chromosomal deletion/transposition.
Although no classification system is perfect, this simple method captures two of the most important prognostic variables of craniosynostosis—the risk of intracranial pressure (ICP) and whether or not there is an associated syndromic diagnosis. Elevations in ICP are directly correlated with the number of fused sutures. Marchac and Renier measured the ICP in 121 patients with craniosynostosis with the help of an epidural sensor. They detected elevated ICP in 42% of patients with multiple-suture involvement and in 7 to 13% of patients with single-suture involvement. They noted a decrease in ICP in patients who underwent cranial release. Similar results have been observed by other authors.
The presence or absence of a syndromic diagnosis greatly affects a patient’s overall prognosis in terms of cognitive and functional capabilities. Most of the genes linked to the various forms of syndromic craniosynostosis (e.g., FGFR-1 to -3) are expressed in multiple tissues during embryogenesis, including neural tissue. The effect of a mutated gene during critical periods of development may, and very likely does, have a more profound effect on neurocognitive development than growth restriction produced by sutural fusion later in childhood. Given the importance of the genetic influences, it is paramount that every child with craniosynostosis undergoes genetic evaluation and testing for commonly associated genetic mutations.
1.4 Diagnosis
Physical examination remains the gold standard for diagnosing craniosynostosis. Most forms of craniosynostosis involve fusion of only a single suture and manifest as a characteristic cranial shape at birth. There are rare forms of craniosynostosis, such as those involving the minor cranial sutures (e.g., frontosphenoidal and squamosal), multiple contiguous or noncontiguous sutures, and latent (postnatal) fusions, that do not fall neatly into the standard phenotypic patterns promulgated by Virchow and are challenging to diagnose by phenotype alone. Most craniofacial surgeons choose to obtain a confirmatory computed tomographic (CT) scan in all patients, but others rely exclusively on the clinical findings for isolated forms of single-suture craniosynostosis. The primary advantage of obtaining a CT before embarking on surgical intervention is to identify any intracranial anomalies (hydrocephalus, arachnoid cysts, etc.) that may also require treatment. In addition, some centers now use virtual surgical planning algorithms that necessitate CT imaging. The disadvantages include the possible need for a general anesthetic and exposure to ionizing radiation. There is evidence that suggests that early exposure to either of these can have possible adverse effects, but the risks are extremely low and must be balanced against the need for clinical information potentially derived from the CT. It is the authors’ current practice to obtain CT imaging on all patients, regardless of the fusion type.
1.5 Functional Aspects
1.5.1 Elevated Intracranial Pressure
The possibility of elevated ICP ranks as the most definitive and objective reason to consider surgical intervention in a patient with craniosynostosis. Prolonged elevated ICP can impair neurocognitive function, lead to blindness, and exacerbate or induce Arnold-Chiari malformation. As noted earlier, the risk of this problem rises geometrically with the number of fused sutures. The most common reason for this finding is a mismatch in shape and volume between the cranium and the brain, but this can be exacerbated by other conditions. Sleep apnea resulting from midfacial retrusion can induce episodic nocturnal elevations in ICP secondary to the dilating effects of hypercapnia on the cerebral vasculature. Another potential cause is venous hypertension resulting from stenosis or complete closure of the sigmoid/jugular sinus complex.
It is important to recognize that although most patients with craniosynostosis do not have elevated ICP, the absence of reliable noninvasive methods of determining ICP (i.e., funduscopic examinations, radiographic changes, and symptoms) and impracticality/morbidity of repeat invasive monitoring make observation a risky proposition. The gold standard for detecting elevated ICP is direct monitoring, which involves placing a transcranial monitor into the cerebral parenchyma or ventricles and monitoring pressure for 12 to 24 hours. This procedure requires an anesthetic and has some risk of intracranial bleeding or cerebrospinal fluid leak. Other methods of assessing ICP such as epidural monitoring and lumbar puncture have fewer complications, but the accuracy of these techniques is questionable. Intracranial pressure readings tend to fluctuate significantly with patient position, activity, blood pressure, and sleep, and the most meaningful results are obtained when patients are monitored for a period of time, usually overnight. Significant elevations (>20 mm Hg) have been considered an absolute indication for intracranial expansion. However, interpretation of the significance of borderline pressure elevations (15–20 mm Hg) has been more problematic, and there is little consensus, even among neurosurgeons.
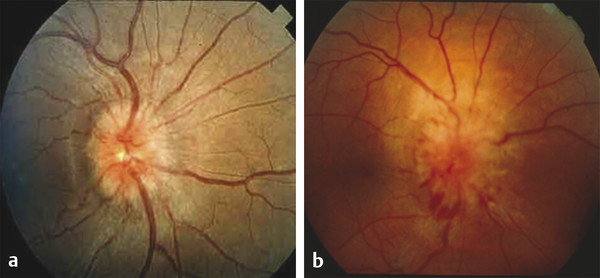
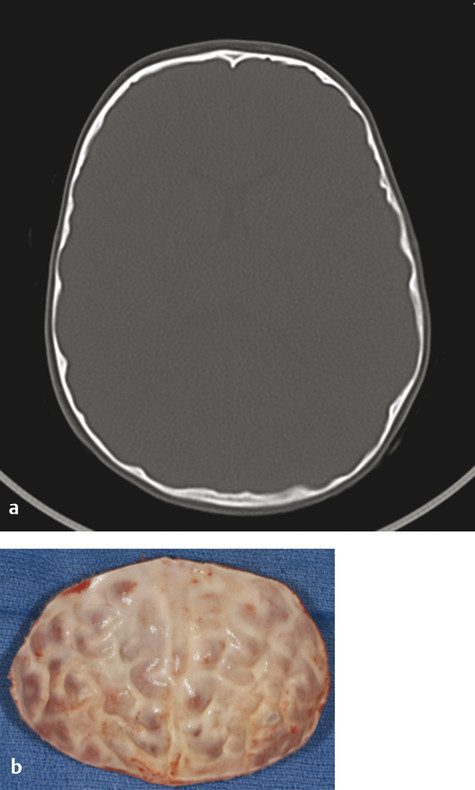
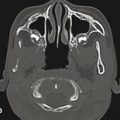
Direct ICP monitoring is invasive and rarely used for routine screening. Moreover, the measurement is only a snapshot in time: a normal pressure measurement early in life does not imply that it will remain so as the brain continues to grow. Consequently, many surgeons resort to noninvasive, but less reliable, indicators of ICP. Conventional clinical symptoms of acute ICP elevation, such as headache, somnolence, and dizziness, are often lacking, even in severely affected children. Papilledema and subsequent optic atrophy are strongly suggestive of elevated ICP but have limited sensitivity in children younger than 8 years. As optic atrophy progresses, the disk becomes pale, the capillaries and hyperemia disappear, and significant secondary arteriolar narrowing occurs (Fig. 1‑3). Reducing ICP can reverse early changes, but more advanced degeneration may be permanent. Radiographic evidence suggestive of elevated ICP includes loss of subdural space, often with effacement of the basal cisterns and vertex sulci, ventricular compression, and scalloping of the cranial endocortex. This latter finding has been termed the “copper beaten” skull and can be visualized on both conventional radiography and CT (Fig. 1‑4). It is a late finding caused by pressure remodeling of the inner table of the skull by the cerebrum. The correlation of this finding with elevation in the ICP has been questioned.
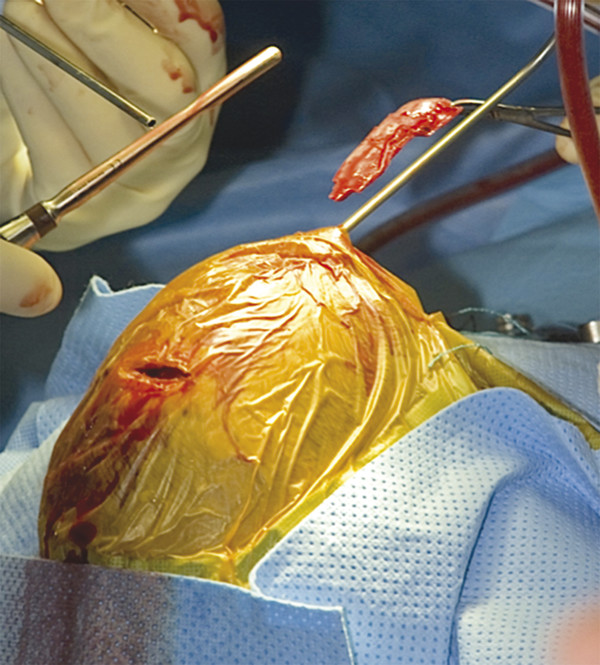
1.5.2 Hydrocephalus
Hydrocephalus is an infrequent finding in patients with craniosynostosis. When it occurs, it is more common in patients with Crouzon’s syndrome but can also occur in other syndromes. It is extremely rare in patients with nonsyndromic, single-suture fusion. Findings of hydrocephalus may include ICP elevation and the presence of enlarged or enlarging ventricles. CT scans provide a reasonable noninvasive method of assessing ventricular size. Interestingly, the authors have observed cranial suture fusion following rapid decompression of hydrocephalus. This postnatal iatrogenic form of craniosynostosis is rare but has been reported.
1.5.3 Mental Impairment
Children with craniosynostosis can exhibit neurocognitive delay and/or learning disabilities. Risk factors include the presence of an associated syndrome or genetic mutation, concurrent ICP elevation, hydrocephalus, prematurity, and family history. Some authorities point to the deleterious effects of early cerebral growth restriction as the primary cause. However, several large investigations have failed to demonstrate that cranial remodeling or volume expansion significantly alters neurocognitive parameters. It is also likely, especially in those with a syndromic diagnosis, that the underlying genetic process that initiated premature sutural fusion also adversely impacted the development of the central nervous system. Patients with single-suture fusion and no associated syndrome generally have near normal intelligence but may exhibit subtle learning disabilities or delayed speech acquisition. In contrast, patients with an associated syndrome have a significantly higher incidence of cognitive delay than in the general population. The degree of cognitive impairment is loosely correlated with the type of syndromic diagnosis, but there is wide variability.
1.5.4 Visual Abnormalities
Ocular anomalies are surprisingly common in patients with craniosynostosis. Hypertelorism, exorbitism, strabismus, and proptosis are observed in many syndromic forms of bilateral coronal craniosynostosis, especially in those with significant midface hypoplasia. These findings are secondary to decreased orbital depth and widening of the ethmoidal air cells and can lead to corneal exposure and ulceration. Hypotelorism and strabismus can be associated with metopic synostosis. Patients with unilateral coronal synostosis have elevation of the lesser and greater sphenoid wings on the side of the fuse suture (harlequin deformity) that results in strabismus and ocular torticollis (head tilt to unfused side) in nearly 80% of affected patients. In addition, the contralateral orbital roof is depressed and 55% of patients have astigmatism. Patients with Saethre–Chotzen syndrome (TWIST mutation) demonstrate upper eyelid ptosis. Many of these manifestations are disfiguring and some can threaten vision. Patients with strabismus or nonconjugate gaze can develop decreased vision (disuse amblyopia) if the visual axis disturbance is not corrected. Strabismus and amblyopia can occurs in up to 40% of patients with syndromic craniosynostosis, but these findings are less common in nonsyndromic patients. Patching of one eye and operative balancing of the extraocular muscles are the mainstays of treatment.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


