Xanthomatoses and Lipoprotein Disorders: Introduction
|
Xanthomas
Xanthomas are plaques or nodules consisting of abnormal lipid deposition and foam cells in skin or in tendons. They do not represent a disease but rather are signs of a variety of lipoprotein disorders. Xanthomas are occasionally seen without an underlying metabolic effect. Xanthomas are thought to develop through several mechanisms. Through scavenger receptors for enhanced low-density lipoprotein (LDL) uptake, macrophages (converted from monocytes) incorporate lipid that has been transported through the capillary walls, thus becoming foam cells. Foam cells can also develop as a result of in situ lipid synthesis by the macrophage. Further, lipid that has been extravasated by the capillaries can also recruit additional foam cells into an already established xanthoma. Extravasated and oxidized LDL recruits foam cells by inducing vascular cellular adhesion molecule and E-selectin.1,2 Local factors such as heat, movement, and friction may increase LDL capillary leakage, and, hence, result in the development of xanthomata.3 These local factors help explain the location of tuberous xanthomas, tendinous xanthomas, and xanthelasmata.
Clinically, xanthomas can be classified as eruptive, tubo-eruptive or tuberous, tendinous, or planar.4 Planar xanthomas include xanthelasma palpebrarum/xanthelasma, xanthoma striatum palmare, and intertriginous xanthomas. There are characteristic clinical phenotypes associated with specific metabolic defects (Tables 135-1 and 135-2 showing older Frederickson classification).
Type of Xanthoma | Genetic Disorders | Secondary Disorders |
|---|---|---|
| Eruptive | Familial lipoprotein lipase deficiency | Obesity |
| ApoC-II deficiency | Cholestasis | |
| ApoA-I and apoA-I/C-III deficiency | Diabetes | |
| Familial hypertriglyceridemia | Medications: retinoids, estrogen therapy, protease inhibitors | |
| Familial hypertriglyceridemia with chylomicronemia | ||
| Tuberous | Familial hypercholesterolemia | Monoclonal gammopathies |
| Familial dysbetalipoproteinemia | Multiple myeloma | |
| Phytosterolemia | Leukemia | |
| Tendinous | Familial hypercholesterolemia | |
| Familial defective apoB | ||
| Familial dysbetalipoproteinemia | ||
| Phytosterolemia | ||
| Cerebrotendinous xanthomatosis | ||
| Planar | ||
| Palmar | Familial dysbetalipoproteinemia | |
| Homozygous apoA-I deficiency | ||
| Intertriginous | Familial homozygous hypercholesterolemia | Cholestasis |
| Diffuse | Monoclonal gammopathies, cholestasis | |
| Familial hypercholesterolemia | ||
| Xanthelasma | Familial dysbetalipoproteinemia | Monoclonal gammopathies |
| Other | ||
| Corneal arcus | Familial hypercholesterolemia | |
| Tonsillar | Tangier disease |
Frederickson Classification | Condition | Defect | Lipoprotein Increase | Type of Xanthoma |
|---|---|---|---|---|
Type I | Familial chylomicronemia | Decreased lipoprotein lipase or altered apoC-II | Chylomicrons (and VLDL) | Eruptive xanthomata |
Type IIa | Familial hypercholesterolemia Heterozygous Homozygous | LDL receptor or apoB deficiency | LDL | Tendon, tuberous, plane xanthomas (xanthelasma and intertriginous) 15% with xanthoma by second decade Xanthomata by age 6 years |
Type IIba | Familial combined hypercholesterolemia | Decreased LDL receptor and increased apoB | LDL and VLDL | Usually absent, but can have xanthomas as in type IIa |
Type III | Familial dysbetalipoproteinemia | Defective apoE-2 synthesis | IDL | Palmar, tuberous, tubo-eruptive xanthoma, xanthelasma |
Type IVb | Familial hypertriglyceridemia (pure) | Increased VLDL production and decreased elimination | VLDL | Rare eruptive xanthomas |
Type V | Mixed hyperlipidemia | Increased VLDL production and decreased LPL | VLDL and chylomicrons | Eruptive xanthomas |
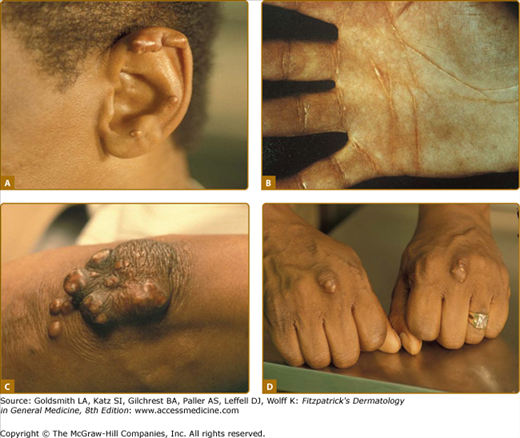
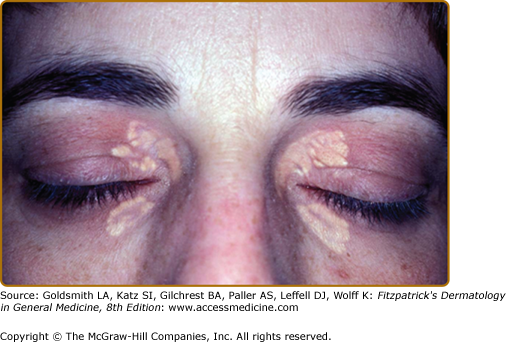
Eruptive xanthomata are multiple, reddish-yellow papules that appear suddenly and are arranged in crops on the extensor surface of the extremities and the buttocks (Figs. 135-1 and 135-2). Tuberous xanthomata are nodules that are frequently localized to the extensor surfaces of the elbows, knees, knuckles, and buttocks (Fig. 135-3). Tendinous xanthomata are firm subcutaneous nodules found in fascia, ligaments, Achilles tendons, or extensor tendons of the hands, knees, and elbows (Fig. 135-4). Planar xanthomata are yellow macules, soft papules, or plaques found commonly on the upper eyelids (xanthelasma palpebrarum), the wrists and palms (xanthoma striatum palmare) (Fig. 135-5), and in intertriginous areas. Xanthelasmas (eFig. 135-5.1) can be found in patients with elevated LDL cholesterol levels, but most often occur in patients with relatively normal lipid levels and the occurrence of these lesions on the eyelids usually not associated with coronary heart disease (CHD). If xanthelasmas cause cosmetic concern, treatment options include surgical excision,42 ablative laser therapy, trichloroacetic acid, or cryotherapy.40
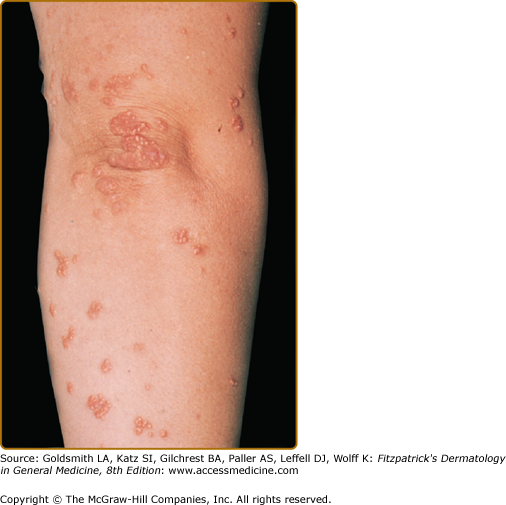
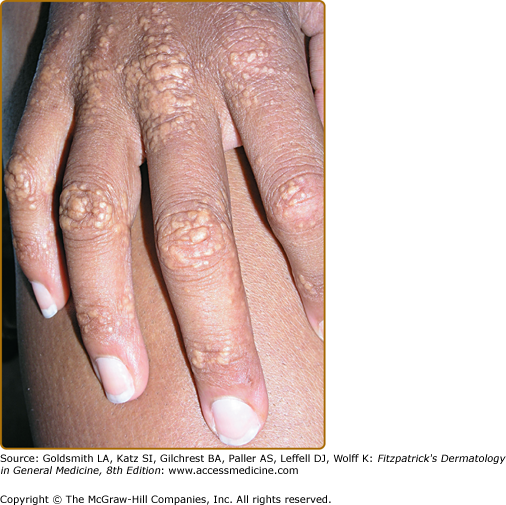
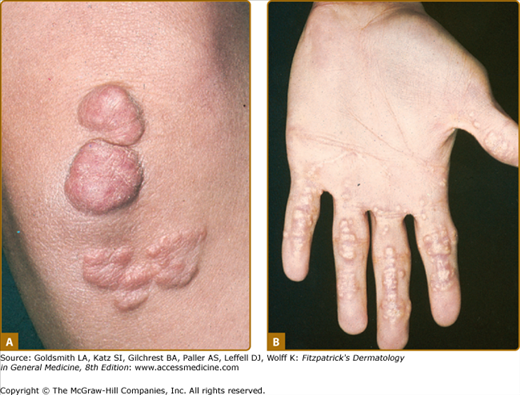
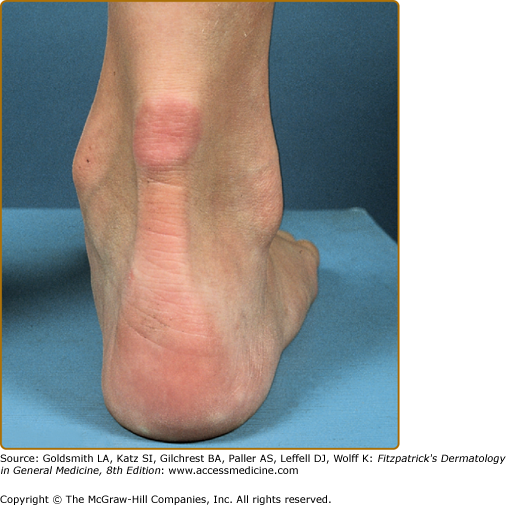
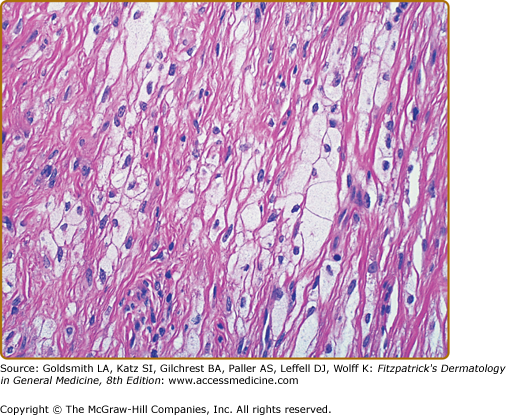
Foam cells are macrophages that contain lipid. These cells characterize xanthomata. Along with foam cells, eruptive xanthomata often consist of lymphoid cells, histiocytes, neutrophils, and, frequently, free lipid in the dermis. Tuberous xanthomata reveal foam cells and cholesterol clefts (Fig. 135-6). Cholesterol is doubly refractile whereas other types of lipid are not. Tendon xanthomata are histopathologically similar to tuberous xanthomata; however, xanthelasmata can be differentiated by their superficial location. Along with the foam cells, xanthelasmata may reveal striated muscle, vellus hair, and/or a thinned epidermis, indicative of the superficial location.5
See Box 135-1.
|
Lipid and Lipoprotein Metabolism
Figure 135-7
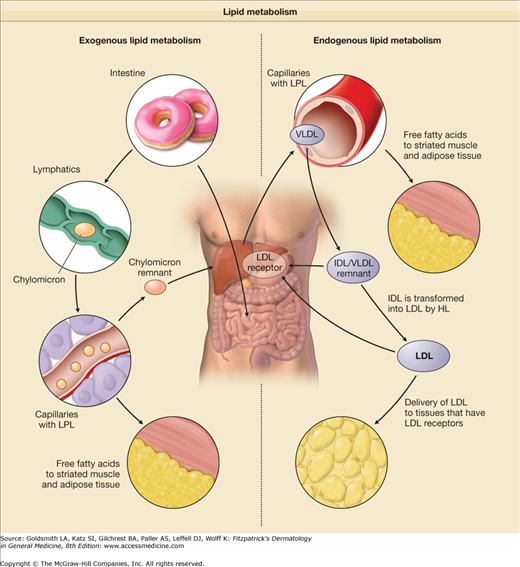
Lipid metabolism. The exogenous pathway of lipid metabolism involves eating foods that contain fat and delivering the fatty acids to tissues. The triglycerides in the intestines are packaged into chylomicrons in the intestinal enterocytes. Capillary lipoprotein lipase (LPL), which requires apolipoprotein IIC (apoC) as a cofactor, hydrolyzes the triglycerides and releases fatty acids to the peripheral tissues. The remaining chylomicron particle is called a chylomicron remnant and is recycled by the liver in a process that requires apoE as a ligand for the LDL receptors in the liver. The endogenous pathway of lipid metabolism involves the hepatic production of very low-density lipoprotein (VLDL) and the delivery of free fatty acids to peripheral tissues. VLDL particles have apoB-100 apoB-100 instead of apoB-48 on the chylomicron. LPL, which requires apoC as a cofactor, hydrolyzes the triglycerides and releases fatty acids to the peripheral tissues. This remnant is called an intermediate-density lipoprotein (IDL). The LDL receptor of the liver removes these remnants by binding to apoE on the remnant. IDL can also be transformed into LDL by hepatic lipase (HL) in the liver. The new particles of LDL can bind to the LDL receptor through apoB-100.
Cholesterol is a waxy substance of molecular weight 387 Da, and is by far the most abundant sterol in plasma. Cholesterol is synthesized in cells in the body, and this source accounts for about 75% of the cholesterol in the bloodstream. Cholesterol serves as a precursor for the bile acids—cholic acid and chenodeoxycholic acid, and steroid hormones including estrogen, testosterone, and cortisol. Cholesterol is also an important constituent of cell membranes. Precursors of cholesterol include lathosterol and desmosterol, which can be measured in plasma or serum and high values indicate increased cholesterol production. Subjects who overproduce cholesterol have elevated absolute levels of these constituents as well as increased values normalized to blood cholesterol levels. Cholesterol production is increased in patients with obesity and metabolic syndrome. There are also patients who have defects in producing the bile acid chenodeoxycholate from cholesterol, leading to cerebrotendinous xanthomatosis.
There are many steps in the cholesterol synthesis pathway from acetate to cholesterol. The rate-limiting enzyme in cholesterol synthesis is 3-hydroxy-3-methylglutaryl CoA reductase or HMG-CoA reductase. Statins competitively inhibit this enzyme, thereby lowering cholesterol production in the body, and decreasing cellular cholesterol synthesis by up to 80%. The cells in the body respond by increasing the level and activity of LDL receptors on their surface, enhancing the clearance of LDL particles from the bloodstream, and lowering LDL cholesterol levels in plasma.3 However, in intestinal cells statins can also increase the amount of cholesterol absorption. Statins are especially effective in subjects who have elevated markers of cholesterol production, and are least effective in patients with elevated plasma markers of absorption.31
Cholesterol is also absorbed in the intestine. About 25% of the cholesterol found in the bloodstream is from dietary sources. The cholesterol found in the intestine is derived from the diet and also made by the liver, secreted into the bile, and reabsorbed. Major dietary sources include eggs, butter, whole milk, and animal fats as found in meat.3 The equivalent substances to cholesterol in plant cells are known as plant sterols (β-sitosterol and campesterol). Almost all of dietary cholesterol and plant sterols are transported into the intestine via the Niemann-Pick C-like protein 1 (NPC1L1) transporter.32–35 This process is blocked about 50% by ezetimibe, a specific inhibitor of NPC1L1.34 Cholesterol in the intestinal cell is either placed onto chylomicrons or high-density lipoproteins (HDL) for entry into the bloodstream, stored in the intestine as either free cholesterol or cholesteryl ester (cholesterol with a fatty acid attached), or transported back out into the intestinal lumen via the action of the two transporters—ATP-binding cassette transporters G5 and G8 (ABCG5 and ABCG8). About 50% of the intestinal cholesterol and more than 95% of the intestinal β-sitosterol and campesterol are transported back out into the intestinal lumen via these ABC transporters. Patients who have defects in ABCG5 and ABCG8 have elevated plasma levels of β-sitosterol as in phytosterolemia, and often develop tendinous xanthomas.5
Cholesterol and triglyceride along with phospholipids are carried in plasma or serum on lipoproteins. Lipoproteins have a surface layer of phospholipids (each phospholipid has two fatty acids attached to it) with the fatty acids directed toward the core of the particle, as well as proteins known as apolipoproteins, and free cholesterol. The hydrophobic components of lipoproteins, namely, cholesteryl ester and triglyceride, are carried within the core of generally spherical lipoprotein particles. Plasma lipoproteins exist in a variety of sizes and densities, ranging from very large triglyceride-rich lipoproteins of density <0.94 g/mL made in the intestine (chylomicrons) to small HDL of density up to 1.21 g/mL.
A model of a spherical lipoprotein particle with proteins (yellow), phospholipids with two fatty acids attached (blue), and free cholesterol (green) on the particle surface, and triglyceride with three fatty acids attached (purple) and cholesteryl ester with one fatty acid attached (green) in the core of the particle is shown in eFig. 135-7.1.
eFigure 135-7.1
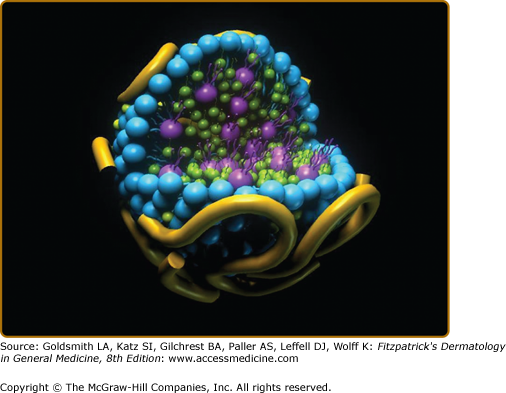
A model of a spherical plasma lipoprotein particle with apoliproteins (yellow), free cholesterol (green), and the polar head groups of the phospholipids (blue) on the surface is shown. Attached to the phospholipid head groups (blue) are two fatty acids oriented toward the center of the lipoprotein particle. In the core of the particle are molecules of cholesteryl ester (green, cholesterol with one fatty acid attached) and triglycerides (glycerol with three fatty acids attached). (Used with permission from Boston Heart Laboratory, Framingham, MA.)
Lipoproteins known as chylomicrons are made in the intestine and vary greatly in molecular weight (50–1,000 × 106 Da) and size (diameter 75–1,200 nm), have a plasma density of <0.93 g/mL, and migrate at the origin on lipoprotein electrophoresis. These particles are very rich in triglyceride (about 85% by weight in the core of the particle) and contain about 3% cholesteryl ester. These particles can also carry significant amounts of fat-soluble vitamins in their core, namely, vitamin A as retinyl palmitate, carotenoids, vitamin D, vitamin E as α- or γ-tocopherol, and vitamin K. On their surface these particles contain about 2% protein, 2% free cholesterol, and 7% phospholipids. The major protein of these particles is known as apolipoprotein (apo) B-48. After chylomicrons are released into the lymph, they pick up apoA-I, apoA-IV and the C apolipoproteins (C-I, C-II, and C-III) on their large surface. The average daily production of apoB-48 in humans is about 2 mg/kg/day.3
Once chylomicrons enter the bloodstream, much of the triglyceride is rapidly removed via the action of lipoprotein lipase (LPL). LPL cleaves the free fatty acids off the glycerol backbone of triglycerides. The apoC-II acts as a cofactor for LPL in this reaction and apoA-V promotes LPL-mediated triglyceride lipolysis. The free fatty acids are taken up by adjacent myocytes and adipocytes. Some free fatty acids bind to albumin and are taken up by fat tissue or transported to a variety of other tissues in the body, including the liver. In the fat the free fatty acids are converted back into triglyceride for long-term energy storage. ApoC-III inhibits this process of lipolysis. When much of the chylomicron triglyceride has been removed, the particles pick up cholesteryl ester from HDL in exchange for triglyceride via the action of cholesteryl ester transfer protein (CETP). They then are much smaller particles and are called chylomicron remnants. In this process of being metabolized to remnants, chylomicrons have lost virtually all of their surface apoA-I, apoA-IV, apoA-V, and C apolipoproteins to HDL, but have retained all of their apoB-48, and have picked apoE from HDL. While the plasma residence time of chylomicron triglyceride is about 5–10 minutes that of chylomicron apoB-48 is about 5 hours.3 Chylomicron remnants are taken up by the liver, a process that is mediated by the binding of apoE to the LDL receptor.3
Very low-density lipoproteins (VLDLs) are made in the liver. They vary in molecular weight (10–80 × 106 Da) and in size (diameter 30–80 nm), have a plasma density of 0.93–1.006 g/mL, and migrate in the prebeta region on lipoprotein electrophoresis. These particles are rich in triglyceride (about 60% by weight in the core of the particle) and contain about 10% cholesteryl ester. On their surface these particles contain about 8% protein, 7% free cholesterol, and 15% phospholipids. Although they resemble chylomicrons, the major protein of these particles is apoB-100, compared to apoB-48 in chylomicrons. Other surface proteins include apoC-I, apoC-II, and apoC-III. In the fed state, the average daily production of VLDL apoB-100 in humans is about 20 mg/kg/day.3 When VLDLs enter the bloodstream, the particles pick up apoE and other apolipoproteins from HDL. Most of the triglycerides in VLDLs are rapidly removed via the action of LPL, similar to intestinal chylomicron particles. In the fat, the free fatty acids are converted back into triglyceride for long-term energy storage. As with chylomicrons, apoC-III inhibits this process of lipolysis. Once VLDL dissociates from LPL, they become intermediate-density lipoproteins (IDLs), which have approximately the same amount of cholesterol and triglyceride. The liver takes up IDL by binding apoE to the LDL receptor and subsequent endocytosis of the IDL. The rest is converted to LDL by hepatic lipase (HL). This conversion process takes about 4–5 hours.3
Low-density lipoproteins (LDLs) are the end product of VLDL catabolism and are mainly produced from the conversion of VLDL to IDL and then to LDL. They are the major cholesterol-transporting lipoproteins in the plasma. LDL have a molecular weight of about 2 × 106 Da, a diameter of 18–25 nm, a plasma density of 1.019–1.063 g/mL, and migrate in the beta region on lipoprotein electrophoresis. These particles are rich in cholesteryl ester (about 40% by weight in the core of the particle) and contain about 5% triglyceride. On their surface, these particles contain about 25% protein, 10% free cholesterol, and 20% phospholipids. The predominant protein of LDL is apoB-100. Occasionally, LDL can contain trace amounts of other surface proteins, namely, apoC-I, apoC-II, apoC-III, and apoE. In the fed state, the average daily conversion of VLDL apoB-100 to LDL apoB-100 in humans takes about 4–5 hours, and is about 12 mg/kg/day. In normal plasma, LDL contains about 60%–70% of the total cholesterol and about 80%–90% of the total apoB. LDL apoB-100 has a plasma residence of about 3.5 days, and is taken by various tissues through the action of the LDL receptor. LDL has been divided into large LDL (density 1.019–1.044 g/mL) and small dense LDL (density 1.044–1.063 g/mL). Small dense LDL is reported to be more atherogenic than large LDL, and its apoB-100 has also has a significantly longer residence time than that of apoB-100 in large LDL.3
Another lipoprotein particle similar to LDL is lipoprotein (a) or Lp(a). This particle is often a small dense LDL particle, with a protein known as apo(a) attached to apoB-100. The apo(a) protein has multiple and variable copies of kringle 4-like domains and one copy of a kringle 5-like domain. These kringles have a high degree of homology with the kringle domains of plasminogen, important for clot lysis. High levels of Lp(a) (>30 mg/dL) are associated with an increased risk of CHD.36,37 Lp(a) is atherogenic because it not only is directly deposited in the artery wall, but also because it may prevent clot lysis by plasminogen. Moreover, Lp(a) serves as an acceptor of oxidized phospholipid from LDL.
High-density lipoproteins (HDLs) (density 1.063–1.21 g/mL) contain about 50% protein, 30% phospholipid, 20% triglyceride, and 5% triglyceride by weight. HDLs show alpha mobility on lipoprotein electrophoresis and have a diameter of about 5.5–12 nm. The major proteins of HDL are apoA-I and apoA-II, and HDL apoA-I has a residence time of about 4.5. HDL participate in reverse cholesterol transport, in which HDL pick up cholesterol from cells from peripheral tissues, and then deliver them to the liver for excretion or transfer it to triglyceride-rich lipoproteins in exchange for triglyceride. In HDL, the enzyme lecithin–cholesterol acyltransferase (LCAT) catalyzes the formation of the cholesterol ester, which are later transferred to lipoproteins via CETP. Decreased HDL cholesterol <40 mg/dL is a significant CHD risk factor.1
Xanthomas are caused by excess deposition of lipid in skin or in tendons, with resultant monocyte migration and uptake of lipid by scavenger receptor on the surface of macrophages (converted from monocytes). Clinically, patients who develop accumulations of chylomicrons and VLDL, remnant lipoproteins, LDL, Lp(a), synthesize excess cholestanol, overabsorb plant sterols, have marked decreases in HDL, or have monoclonal gammopathies or multiple myeloma can develop xanthomas.4–60
Patients that have defects in the catabolism of triglycerides on chylomicrons and VLDL can develop severe hypertriglyceridemia, which can lead to eruptive xanthomas. Patients that have defects in the clearance of chylomicron and VLDL remnant particles can develop combined hyperlipidemia and tubo-eruptive xanthomas. Such xanthomas can also be observed in marked HDL deficiency due to lack of HDL apoA-I production. Patients that have defects in the clearance of LDL can develop very high LDL cholesterol levels and may develop tendinous xanthomas. Such xanthomas can also be seen in patients who synthesize excess cholestanol or overabsorb pant sterols. Both cholestanol and plant sterols (β-sitosterol and campesterol) are then carried in excess on LDL particles.
Xanthomas are uncommonly seen in common familial lipoprotein disorders associated with premature CHD.
Common genetic lipoprotein disorders associated with premature coronary heart disease include familial Lp(a) excess (Lp(a) >30 mg/dL), seen in about 20% of families with premature CHD, familial combined hyperlipidemia (LDL cholesterol >160 mg/dL and triglyceride >150 mg/dL) (15% of families) and familial dyslipidemia (triglycerides >150 mg/dL and HDL cholesterol <40 mg/dL) (15% of families), and familial hypoalphalipoproteinemia (isolated low HDL cholesterol <40 mg/dL) (5% of families).38 Patients with markedly elevated Lp(a) levels or with markedly elevated levels of LDL cholesterol and triglycerides due to familial combined hyperlipidemia occasionally develop tendinous or tubo-eruptive xanthomas. In addition, patients with elevated levels of triglycerides (>400 mg/dL) and low HDL cholesterol (<40 mg/dL) or marked HDL deficiency alone may occasionally develop tubo-eruptive xanthomas. Tangier disease is a rare, autosomal-recessive condition that often leads to CHD, but is not typically associated with xanthomas.
 Lipoprotein (a) levels [mainly LDL with apo(a) attached] are in large part determined by the number of apo(a) isoforms, which are inherited. A decreased number of kringle 4-like repeats results in less intrahepatic degradation of apo(a) and more secretion. Most patients with familial Lp(a) excess have decreased numbers of kringle 4 repeats. Familial lipoprotein(a) excess is found in about 20% of familial premature coronary heart disease (CHD).38 The metabolism of apo(a) requires further elucidation. In our view, apo(a) attaches itself to VLDL apoB-100, and then remains with VLDL as it is converted to LDL, or if the VLDL is catabolized directly, the apo(a) is detached and recombines with a newly formed VLDL particle, or is catabolized. Lp(a) excess is associated with both increased apo(a) and apoB-100 secretion into plasma Lp(a), as well as delayed clearance of apo(a), especially in patients with elevated LDL.39 Lp(a) is measured by using immunoassays specific for apo(a). Elevated levels of Lp(a) have been shown to be an independent predictor of CHD. Elevated Lp(a) levels can be lowered by using niacin at a dose of 2 g/day.40 Clinical trials currently underway with niacin will test the hypothesis whether lowering elevated Lp(a) will reduce CHD risk. Patients with markedly elevated Lp(a) levels may occasionally develop tendinous or tubo-eruptive xanthomas.
Lipoprotein (a) levels [mainly LDL with apo(a) attached] are in large part determined by the number of apo(a) isoforms, which are inherited. A decreased number of kringle 4-like repeats results in less intrahepatic degradation of apo(a) and more secretion. Most patients with familial Lp(a) excess have decreased numbers of kringle 4 repeats. Familial lipoprotein(a) excess is found in about 20% of familial premature coronary heart disease (CHD).38 The metabolism of apo(a) requires further elucidation. In our view, apo(a) attaches itself to VLDL apoB-100, and then remains with VLDL as it is converted to LDL, or if the VLDL is catabolized directly, the apo(a) is detached and recombines with a newly formed VLDL particle, or is catabolized. Lp(a) excess is associated with both increased apo(a) and apoB-100 secretion into plasma Lp(a), as well as delayed clearance of apo(a), especially in patients with elevated LDL.39 Lp(a) is measured by using immunoassays specific for apo(a). Elevated levels of Lp(a) have been shown to be an independent predictor of CHD. Elevated Lp(a) levels can be lowered by using niacin at a dose of 2 g/day.40 Clinical trials currently underway with niacin will test the hypothesis whether lowering elevated Lp(a) will reduce CHD risk. Patients with markedly elevated Lp(a) levels may occasionally develop tendinous or tubo-eruptive xanthomas.
 The most common familial cause of elevated LDL cholesterol is known as familial combined hyperlipidemia, found in about 15% of patients with premature CHD.38
The most common familial cause of elevated LDL cholesterol is known as familial combined hyperlipidemia, found in about 15% of patients with premature CHD.38
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


