Vascular Tumors and Malformations
Edward P. Conrad
VASCULAR TUMORS
Infantile Hemangiomas
I. BACKGROUND
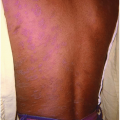
Infantile hemangiomas are the most common softtissue tumors of infancy, occurring in approximately 10% of the population (Fig. 45-1). The etiology is unknown, but identifiable risk factors include female sex, premature birth, low birth weight, and fair skin. They consist of rapidly dividing benign endothelial cells.
II. CLINICAL PRESENTATION
Infantile hemangiomas typically present during the first few weeks to months of life. The morphology depends on the depth within the skin. Superficial hemangiomas present as well-demarcated, finely lobulated, bright red papules/plaques, while deep hemangiomas present as a soft, ill-defined subcutaneous faint blue masses. There are often components of both. They follow a characteristic growth pattern of rapid growth in the first few weeks, followed by a period of slower growth between 6 and 12 months of age, followed by a slow involution. The exact time frame for growth and involution is difficult to predict. In general, 30% involute by the age of 3, and an additional 10% involute each additional year of life (i.e., 40% by age 4, 50% by age 5, and 90% by age 9).
III. WORKUP
Most infantile hemangiomas do not require any workup. Doppler ultrasound can help confirm the diagnosis if not evident clinically. Extracutaneous disease is associated with certain anatomical sites and therefore may require some additional workup. Large facial hemangiomas may be associated with other congenital anomalies in PHACES syndrome (posterior fossa defects, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, eye abnormalities, and sternal/supra-abdominal clefting). These patients should be sent for ophthalmologic examination, echocardiogram, and a magnetic resonance imaging (MRI)/magnetic resonance angiography of the head and neck (Table 45-1). Lumbosacral hemangiomas may be associated with genitourinary anomalies such as an imperforate anus, renal anomalies, and underlying spinal cord anomalies. High-resolution ultrasound or MRI should be done to look for spinal cord involvement. If numerous lesions are present at birth, there is an increased risk of visceral lesions, particularly liver and gastrointestinal tract. Liver ultrasound and stool guaiac should be performed.
IV. TREATMENT
The prognosis of the vast majority of infantile hemangiomas is excellent. Spontaneous involution is the typical outcome, so most infantile hemangiomas do not need any treatment. There are several exceptions.
Involution of the vascular component does not always result in completely normal skin, as larger, exophytic hemangiomas leave residual fibrofatty scarring that can be disfiguring. Astigmatism, strabismus, and amblyopia can result from periocular skin involvement. Hemangiomas involving the mandible, chin, and upper neck may be associated with airway involvement and respiratory distress. Ulceration is the most common complication, which can result in scarring, pain, and potentially infection. Up until recently, oral and intralesional corticosteroids have been the mainstay of treatment. However, the use of systemic β-blockers has been proven to be a potentially safer and more effective treatment alternative. Over 90% of patients have dramatic reduction in the size of their hemangiomas as early as 1 to 2 weeks after initiation of treatment. There are currently no consensus guidelines regarding the use of propranolol for the treatment of
hemangiomas, and studies to determine the optimal dosing and monitoring are ongoing. Topical β-blockers such as timolol have also been found to be of use. Vincristine, recombinant interferon-α, pulsed dye laser, Nd:YAG, and surgical excision are other treatment options for treatment-resistant lesions.
hemangiomas, and studies to determine the optimal dosing and monitoring are ongoing. Topical β-blockers such as timolol have also been found to be of use. Vincristine, recombinant interferon-α, pulsed dye laser, Nd:YAG, and surgical excision are other treatment options for treatment-resistant lesions.
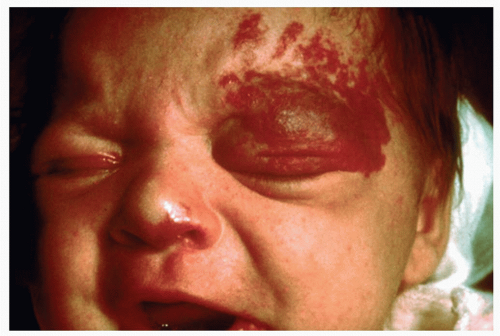 Figure 45-1. Infantile hemangioma. (From Rubin E, Farber JL. Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.) |
TABLE 45-1 Hemangiomas with Suspected Congenital Anomalies | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Congenital Hemangiomas
I. BACKGROUND
Congenital hemangiomas are much less common than infantile hemangiomas. They are present at birth and classified as either rapidly involuting congenital hemangioma (RICH) or noninvoluting congenital hemangioma (NICH), which never involutes at all.
II. CLINICAL PRESENTATION
They typically present as pink to violaceous masses with overlying coarse telangiectasias, surrounded by a pale rim. RICHs are characterized by rapid involution over the first year of life, while NICHs grow proportionately with the child.
III. WORKUP
Doppler ultrasound may be used to confirm the diagnosis.
IV. TREATMENT
Surgical excision.
Kaposiform Hemangioendothelioma
I. BACKGROUND
Kaposiform hemangioendotheliomas (KHs) are rare vascular tumors that typically present during the first months to 1 year of life. They are one of the major causes (in addition to tufted angiomas) of Kasabach-Merritt phenomenon (KMP), a coagulopathy characterized by thrombocytopenia.
II. CLINICAL PRESENTATION
KHs present as rapidly enlarging, subcutaneous, ecchymotic masses. They often become more indurated and enlarged when the coagulopathy develops. Residual masses often persist even after the coagulopathy resides.
III. WORKUP
Complete blood count (CBC) should be closely monitored, as thrombocytopenia is a common complication.
IV. TREATMENT
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


