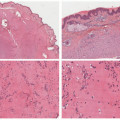
area of pallor or telangiectasia; initially, they may resemble a bruise. Hemangiomas may occur anywhere on the body but may have a predilection for the head and neck. Hemangiomas grow rapidly over the first 5 to 7 weeks of life, ultimately growth stabilizes, and IH involute or regress slowly over the course of years.4 There are several clinical subtypes, including superficial, deep, mixed, and IH with minimal or arrested growth. All have a typical clinical appearance in the skin. Superficial IH are well-demarcated bright red vascular plaques. Deep IH are bluish-hued subcutaneous nodules. Mixed IH demonstrate both a red superficial component and a deeper underlying bluish tumor. IH with minimal or arrested growth tend to favor the lower body and are often flat or telangiectatic in their appearance, with proliferation occurring only in <20% of the lesion, most often at the periphery (Figure 25-1A-D clinical subtypes). IH may be solitary or multifocal in their presentation. When they appear to occupy a region or territory of the body, for example large facial hemangiomas on the frontotemporal area, they may be associated with underlying structural anomalies as seen in Posterior fossa anomalies, large facial Hemangioma, Arterial anomalies, Cardiac anomalies, Eye anomalies (PHACE).5
TABLE 25-1. Classification of vascular anomalies: basic overview2 (ISSVA, 2014) | ||||||
|---|---|---|---|---|---|---|
| ||||||
TABLE 25-2. Vascular tumors (ISSVA, 2014) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
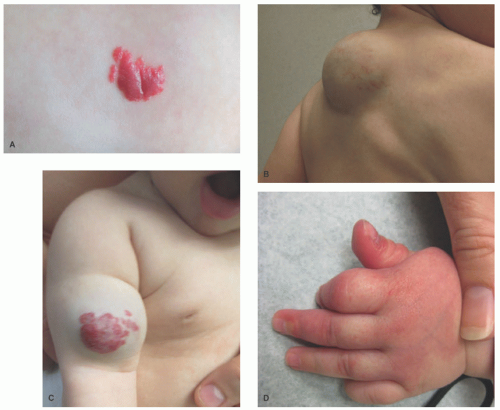 FIGURE 25-1. A, Superficial infantile hemangioma. B, Deep infantile hemangioma. C, Mixed, superficial, and deep infantile hemangioma. D, Infantile hemangioma with minimal or arrested growth. |
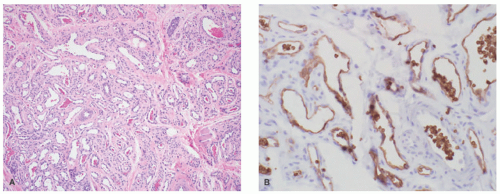 FIGURE 25-2. Infantile hemangioma (IH). A, Lobulated clusters of small, thin-walled vessels with minimal intervening stroma are present in the dermis (H&E-stained section, 100× magnification). B, Glucose transporter protein isoform 1 immunostain, with a positive labeling of the endothelium of lesional vessels in IH. |
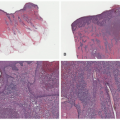
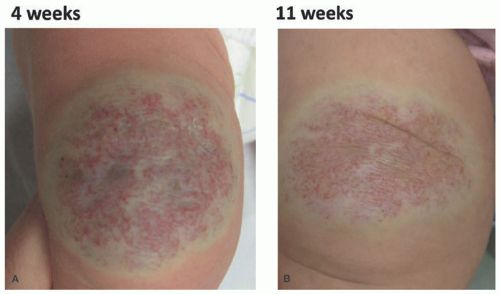 FIGURE 25-3. Rapidly involuting congenital hemangioma of the left leg (A) at 4 weeks (B) at 11 weeks. |
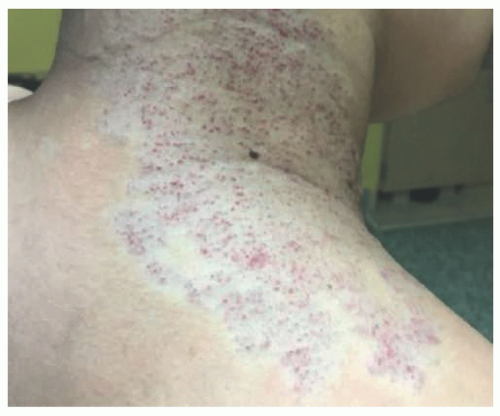 FIGURE 25-4. Noninvoluting congenital hemangioma of the neck. |
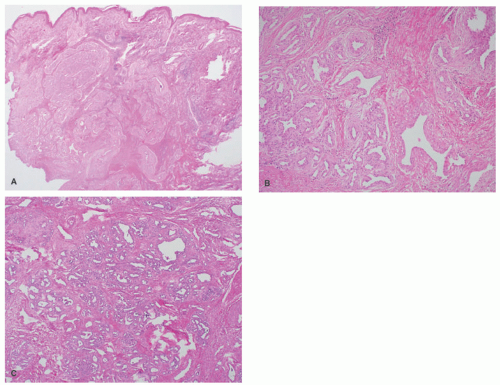 FIGURE 25-5. Histopathology of congenital hemangiomas. A, Rapidly involuting congenital hemangioma: Similar-sized lobules of capillaries fill and expand the dermis (H&E, 20× magnification). B, Partially involuting congenital hemangioma: Vascular lobules with intervening stromal fibrosis indicative of partial involution (100× magnification). C, Noninvoluting congenital hemangioma: Variably sized capillaries with hyperchromatic, occasionally hobnailing, endothelial cells (40× magnification). |
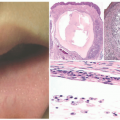
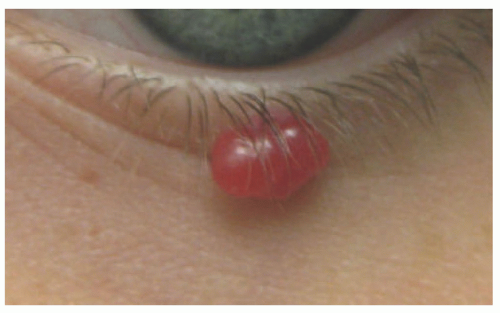 FIGURE 25-6. Pyogenic granuloma on the lower eyelid margin. |
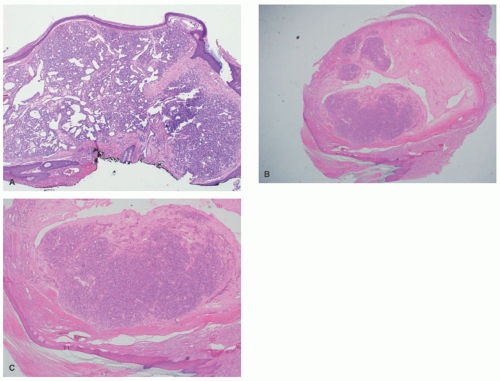 FIGURE 25-7. Pyogenic granuloma (PG). A, An epidermal collarette is seen surrounding an upper dermal proliferation of small, thin-walled vessels arranged in lobules and separated by thin fibrous septae (H&E-stained section, 40× magnification). B and C, Intravascular PG: clusters of small vessels with a lobulated architecture filling a dilated, thin-walled vessel (20× and 40× magnification, respectively). |
demonstrates increased fibrosis and diminished vascular lobules. Deep dermal or subcutaneous presentations of PG are not uncommon and may be intravascular. Intravascular PG presents within the lumen of a vein or artery. Intravenous PG was originally described by Cooper et al. in 1979,18 as an intraluminal polyp comprised by lobules of capillaries lined by flattened or rounded endothelial cells (Figure 25-7B). A surrounding fibrous stroma is also usually observed.19,20
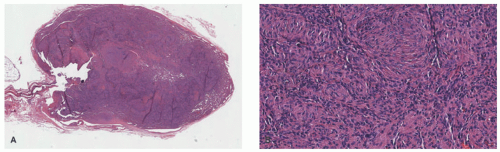 FIGURE 25-8. Spindle cell hemangioma. A, Well-circumscribed tumor with dense cellularity centrally and dilated thin-walled vessels peripherally (20× magnification). B, Sheets of spindled cells that may resemble nodular Kaposi’s sarcoma (200× magnification). Digital slides courtesy of Path Presenter.com. |
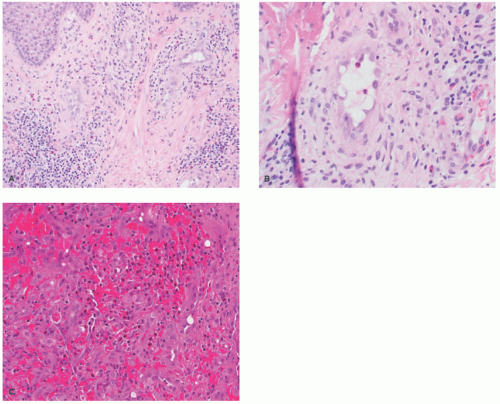 FIGURE 25-9. Epithelioid hemangioma (EH). A and B, Angiolymphoid hyperplasia with eosinophilia-type. Small vessels with epithelioid endothelial cells with vacuolated cytoplasm, and surrounding inflammation rich in eosinophils (100× and 200× magnification). C, Cellular EH variant that may mimic epithelioid hemangioendothelioma or angiosarcoma (200× magnification). |
lymphoid follicles) and a lesser vascular element. More cellular EH types may mimic malignant tumors such as epithelioid hemangioendothelioma (EHE) or even epithelioid angiosarcoma (AS) (Figure 25-9C). Immunohistochemistry and molecular studies such as fluorescent in situ hybridization (FISH) can be helpful in ruling in these malignant tumors, because 90% of EHE demonstrate a characteristic WWTR1-CAMTA1 fusion, which results from a t(1;3)(p36;q25) translocation,29 and many ASs demonstrate MYC amplification on FISH and/or MYC-positivity on immunohistochemistry.30
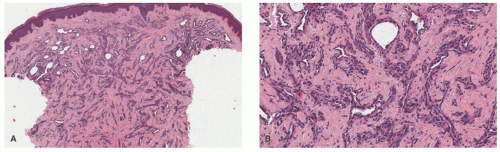 FIGURE 25-10. Microvenular hemangioma. A and B, Interstitially arrayed small, thin-walled vessels dissecting between dermal collagen fibers (40× and 100× magnification). Digital slides courtesy of Path Presenter.com. |
histopathologic features and thus it is felt that they may exist together on a spectrum. On one end, TAs are considered benign and tend to be smaller in size, whereas KHE is locally aggressive and potentially life threatening. Both may be associated with a consumptive coagulopathy known as Kasabach Merritt phenomenon (KMP), which is the main source of mortality in the setting of KHE. This phenomenon does not occur in the more common entity of IH. KMP occurs because of platelet trapping within the aberrant vessels of these two unique tumors. In extensive, infiltrative lesions, more commonly with KHE, this leads to profound thrombocytopenia, hypofibrinogenemia, elevated D-dimer, and prolonged prothrombin time and activated partial thromboplastin time. Infants with KMP are at risk for life-threatening bleeding/hemorrhage.36
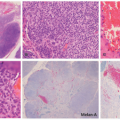
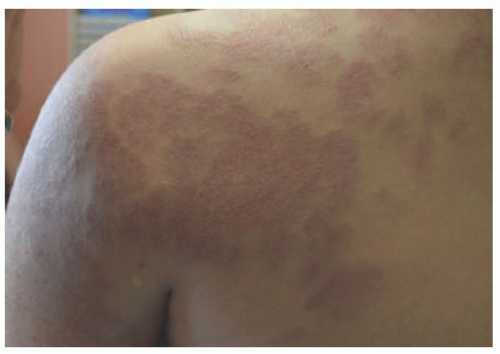 FIGURE 25-11. Tufted angioma on the left upper extremity in a 10-year-old female. |
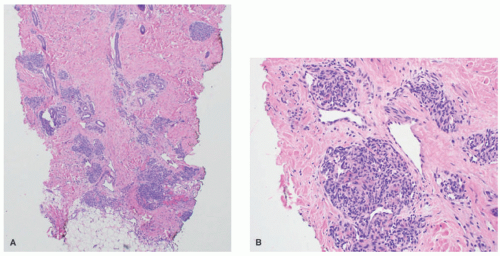 FIGURE 25-12. Tufted angioma. A, Rounded lobules of small, thin-walled vessels in the deep dermis, with minimal intervening stromal changes and absence of subcutaneous extension (H&E-stained section, 40× magnification). B, Crescentic lymphatic channels encircle individual lobules (H&E-stained section, 200× magnification). |
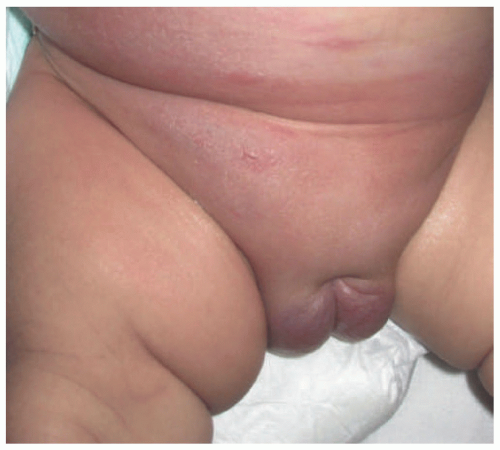 FIGURE 25-13. Kaposiform hemangioendothelioma in the inguinal region of an infant. Photo courtesy of Dr. Kristen Hook. |
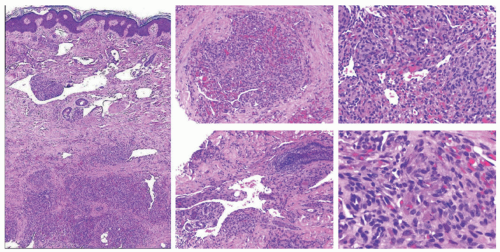 FIGURE 25-14. Kaposiform hemangioendothelioma. Tumor with lobulated zones that resemble those of tufted angioma, but also with increased interlobular vessels in a dissecting pattern similar to that of Kaposi’s sarcoma. |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


