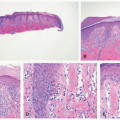
contours, significant variability in diameters, and disorganized packing into larger fibers.
TABLE 9-1. Categorization and features of Ehlers-Danlos syndrome | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 9-2. Pediatric findings in Ehlers-Danlos syndrome | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
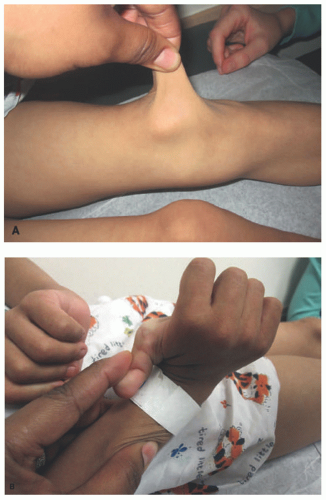 FIGURE 9-1. Ehlers-Danlos syndrome. Skin hyperextensibility (A) and joint hypermobility (B). |
joint hypermobility syndrome should be easily excluded by the lack of cutaneous findings of hypermobile EDS and, if uncertain, by the strict application of diagnostic criteria.
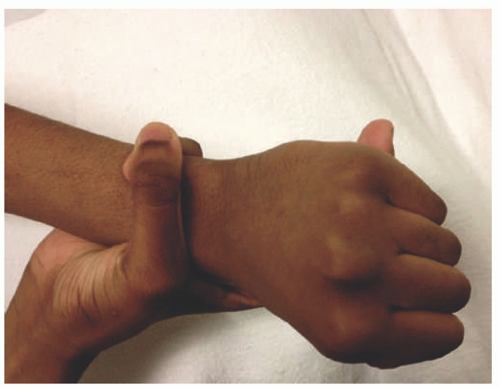 FIGURE 9-2. Marfan syndrome. Long fingers result in the thumb sign, in which the thumb protrudes from a closed fist, and the wrist sign, in which the thumb and fifth finger overlap when encircling the opposite wrist. Reprinted with permission from Bitterman AD, Sponseller PD. Marfan syndrome: a clinical update. J Am Acad Orthop Surg. 2017;25(9):603-609, Figure 2. |
FBN2 cause congenital contractural arachnodactyly (CCA), which is characterized by contractures, arachnodactyly, scoliosis, and crumpled ears. These findings can be seen in neonatal MFS, but aortic root involvement and valvular disease is not typical of CCA.28 MFS-spectrum disorders (mitral valve prolapse syndrome, ectopia lentis syndrome, and MASS) can all be diagnosed under the current guidelines.14
TABLE 9-3. Causes and features of cutis laxa | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
disease is caused by mutations in elastin support proteins (FBLN4 and FBLN5), the TGF-β pathway (LTBP4), vesicular ATPase (ATP6V0A2, ATP6V1E1, ATP6V1A), vesicular trafficking proteins (GORAB, RIN2), and mitochondrial components (PYCR1, ALDH18A1). Occipital horn syndrome, previously categorized as a subtype of EDS, is caused by an X-linked defect in cooper transport (ATP7A). Many other congenital syndromes also feature hypoelastic skin.
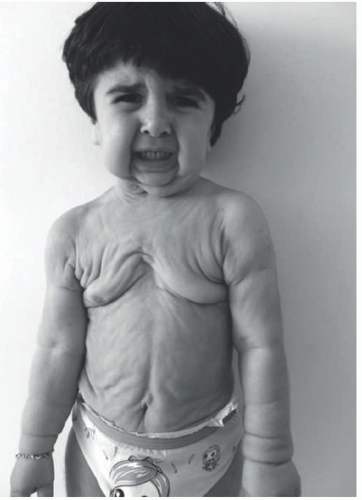 FIGURE 9-3. Cutis laxa. Pendulous skin folds confer a progeroid appearance. Reprinted with permission from Duz MB, Kirat E, Coucke PJ, et al. A novel case of autosomal dominant cutis laxa in a consanguineous family: report and literature review. Clin Dysmorphol. 2017;26(3):142-147. |
underlying bone and meninges are not uncommon. There is no single cause of ACC, which can be categorized on the basis of the number and location of lesions and associated clinical features (Table 9-4). The estimated incidence of ACC is approximately 3 in 10 000 births with a predominance in females.48,49
TABLE 9-4. Causes of aplasia cutis congenita | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
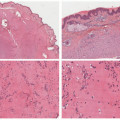
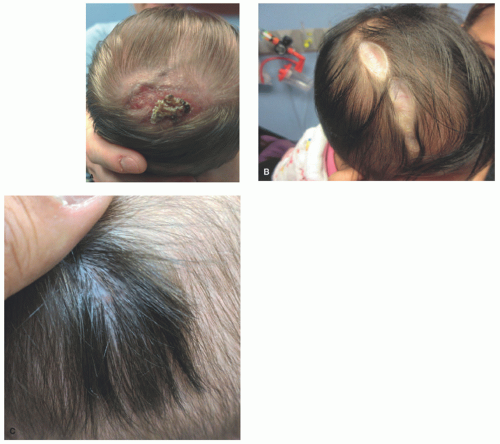 FIGURE 9-4. Aplasia cutis congenital. A, A crusted, healing erosion on the vertex of the scalp in a newborn infant. B, Extensive, well-healed aplasia cutis with resultant atrophic plaques. C, Aplasia cutis with hair collar sign—a rim of dark hair that may indicate underlying skull or central nervous system defects. |
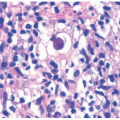
epidermis overlies an edematous dermis with very loose connective tissue.
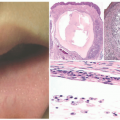
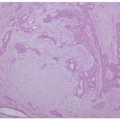
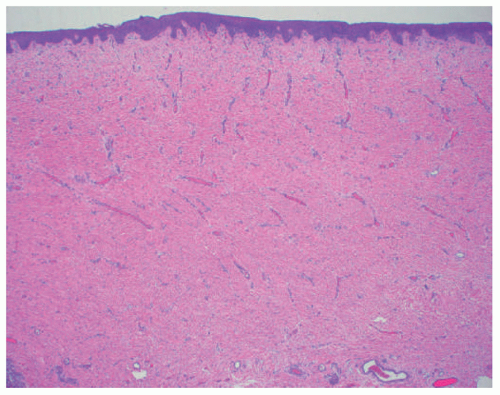 FIGURE 9-5. Aplasia cutis congenital. Thin epidermis overlies a dermal scar that lacks adnexal structures.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|