Genetic Disorders of DNA Repair Mechanisms
Jennifer A. Day
Adnan Mir
XERODERMA PIGMENTOSUM
Definition and Epidemiology
Xeroderma pigmentosum (XP) is a group of genetically inherited defects caused by mutations in genes responsible for nucleotide excision repair (NER), leading to increased malignancy risk.1,2,3 The reported incidence ranges from 1 in 20 000 to 40 000 live births in Japan to 1 in 250 000 to 1 000 000 in the United States and Europe.4 The prevalence of subtypes varies geographically, with the complementation group XPA being the most common in Japan, and XPC being the most common in the United States and Europe. The Navajo populations in the Southwestern United States manifest an uncommon XPA mutation that has been found in both homozygote and heterozygote states because of a founder effect.5,6
Etiology
XP results from mutation of genes in the NER pathway, XPA-XPG (Table 11-1). The resultant abnormal repair of ultraviolet (UV) radiation-induced DNA damage is caused by inability to repair pyrimidine dimers, leading to the accumulation of UV signature mutations. XP Variant (XPV) is a related disorder resulting from a defect in a postreplication repair mechanism carried out by a protein encoded by the DNA polymerase ç gene.7
Clinical Presentation
The clinical manifestations of XP vary by clinical subtype, which are determined by complementation groups (Table 11-1).8 Generally, complementation groups are divided among those associated with severe photosensitivity and neurologic complications (XPA, XPB, XPD, XPF, and XPG) and those with no neurologic complications (XPC, XPE, and XPV).9
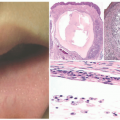
Early cutaneous manifestations include photosensitivity, ephelides, lentigines, and poikiloderma (Figure 11-1).6,7 Photosensitivity manifests as exaggerated sunburn, blisters, and persistent erythema, often after minimal UV exposure.5 Later findings include premature photoaging and xerosis.1 The hallmark cutaneous manifestation is the marked predisposition for premature cutaneous malignancy. This increased risk manifests in childhood and adolescence.
The median age for the initial diagnosis of nonmelanoma skin cancer in XP patients is 9 years, with patients under 20 years old having a 10 000-fold increased risk of nonmelanoma skin cancer (typically basal cell carcinoma [BCC] or squamous cell carcinoma [SCC], respectively).10 The median age for the initial diagnosis of melanoma is 22 years, with a risk 2000 times the typical population.11,12 There are reports of additional nonmelanoma skin cancers in XP patients, including atypical fibroxanthoma, malignant fibrous histiocytoma, fibrosarcoma, and angiosarcoma.12
The median age for the initial diagnosis of nonmelanoma skin cancer in XP patients is 9 years, with patients under 20 years old having a 10 000-fold increased risk of nonmelanoma skin cancer (typically basal cell carcinoma [BCC] or squamous cell carcinoma [SCC], respectively).10 The median age for the initial diagnosis of melanoma is 22 years, with a risk 2000 times the typical population.11,12 There are reports of additional nonmelanoma skin cancers in XP patients, including atypical fibroxanthoma, malignant fibrous histiocytoma, fibrosarcoma, and angiosarcoma.12
TABLE 11-1. Xeroderma pigmentosum complementation groups | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
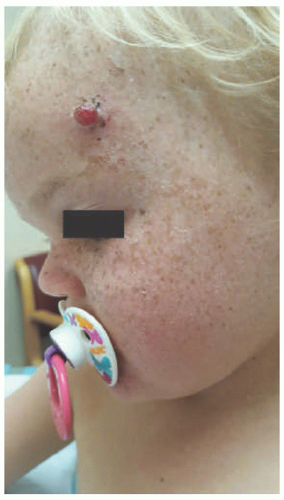 FIGURE 11-1. Early-onset photo damage in a child with xeroderma pigmentosa, with photodistributed actinic damage, solar lentigos, and an early squamous cell cancer (Courtesy Barrett Zlotoff, MD). |
Ocular manifestations are reported in 40% of XP patients.2,13 Findings can include photophobia, conjunctivitis, ectropion, and symblepharon (adhesion of the palpebral conjunctiva to the bulbar conjunctiva). Corneal involvement may lead to blindness. Ocular neoplasms occur in about 11% of XP patients, including epithelioma, BCC, SCC, and melanoma.11 Neurologic findings are highly variable depending on the complementation group, with the highest rate in patients with XPD.14 Manifestations may include developmental delay, intellectual impairment, sensorineural hearing loss, hyporeflexia, and ataxia.15 A rare XP phenotype is De Sanctis-Cacchione syndrome, in which patients exhibit severe mental retardation, deafness, ataxia, and paralysis.16 Notably, neurologic manifestations are absent in XPV.17 The risk for internal malignancy, most notably brain cancer, is also increased in XP patients.2,11,18 Mortality typically occurs in the third or fourth decade because of neurologic complications, metastatic SCC, or melanoma.10,15
Histologic Findings
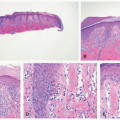
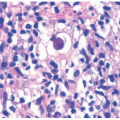
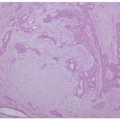
Early histologic changes include telangiectasia and pigmentary changes such as prominent epidermal melanin and pigmentary incontinence.19 Solar elastosis develops within a few years of life. As tumors develop (Figure 11-2A-C), lesional specimens may demonstrate findings of angiomas, actinic keratoses, keratoacanthomas, BCC, SCC, melanoma, and atypical fibroxanthoma.12
Differential Diagnosis
The differential diagnosis for photosensitivity seen in XP includes Cockayne syndrome (CS), UV-sensitive syndrome, cerebro-oculo-facio-skeletal syndrome, and erythropoietic protoporphyria. The differential diagnosis for photosensitive disorders with predisposition toward malignancy includes Bloom and Rothmund-Thomson syndromes (RTS).
CAPSULE SUMMARY
XERODERMA PIGMENTOSUM
XP is a rare group of genetic disorders resulting from defective NER, leading to markedly increased risk for cutaneous malignancy at an early age. Specific histologic findings include those related to actinic damage including telangiectasias and pigmentary change. Patients develop actinic keratoses, BCC, SCC, melanoma, and other cutaneous neoplasms.
COCKAYNE SYNDROME
Definition and Epidemiology
CS is a rare group of heritable defects due to mutations in genes involved with NER, leading to photosensitivity.20 The estimated incidence ranges from 1 in 1.8 to 2.7 million in Europe.21 It is reportedly more prevalent in Canada, Japan, and Middle Eastern and Western Asian countries.21
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


