Cutaneous Mucinosis, Deposits, and Cysts
Shivani S. Patel
Anna L. Grossberg
Mark R. Wick
Deposition Disorders of the Skin
Several cutaneous deposition disorders may affect children. These can be grouped into conditions that feature excess stromal mucin or mucopolysaccharide in the corium, forms of amyloidosis, and deposits of minerals or mineral salts.
Mucin Deposition Disorders and Mucopolysaccharidoses
MYXEDEMA AND PRETIBIAL MYXEDEMA
Etiology, Definition and Epidemiology
Myxedema and pretibial myxedema are both rare in children. The former is associated with congenital hypothyroidism, which has largely been eliminated as a medical problem in the modern world because of common screening studies for thyroid function in newborns. Pretibial myxedema is a thyroid dermopathy characteristic of Graves’ disease.
The incidence is 1 per 100 000 and occurs in approximately 4% of patients with Graves’ disease.1 There is a female predominance. Pretibial myxedema is rare in children but has been reported in adolescents.
Clinical Presentation and Pathogenesis
Myxedema classically presents on the shins, but has also been reported on the arms, shoulders, and upper back at the sites of trauma.1,2 There is usually symmetric, nonpitting edema resembling an orange peel both in appearance and texture because of hair follicle accentuation (Figures 15-1 and 15-2). Up to 40% of patients will also develop overlying erythematous or skin-colored plaques and nodules. There is usually no associated pain or pruritus.1 This manifestation usually appears 1 to 2 years after a diagnosis of Graves’ disease and is more common in patients with associated ophthalmopathy.
Pathophysiology is poorly understood but may involve an overexpression of the thyrotropin receptor serving as a nonspecific antigen on fibroblasts, leading to Interleukin-1 and transforming growth factor-beta release. Cytokine production then stimulates a synthesis of glycosaminoglycans by dermal fibroblasts.1,2,3 The skin becomes thickened and “doughy” because of this excess production of stromal mucin by dermal fibroblasts. Studies also demonstrate an
upregulation of insulin-like growth factor (IGF-1), leading to fibroblast activation and T-cell response in the dermis. Pretibial myxedema has been reported in skin harvested from other sites and grafted to the legs, suggesting a role for dependent position and mechanical stress.3
upregulation of insulin-like growth factor (IGF-1), leading to fibroblast activation and T-cell response in the dermis. Pretibial myxedema has been reported in skin harvested from other sites and grafted to the legs, suggesting a role for dependent position and mechanical stress.3
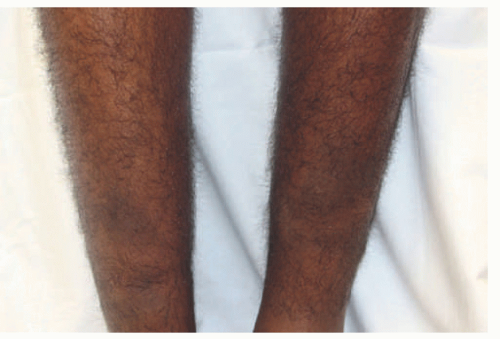 FIGURE 15-1. Pretibial myxedema of the shins in a young boy with thyroid disease. |
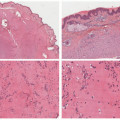
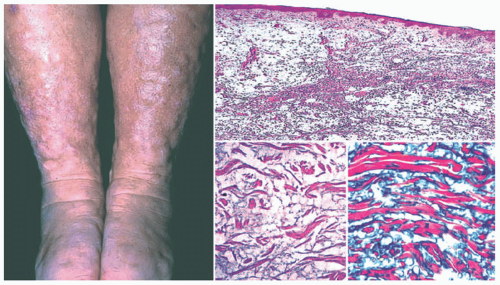 FIGURE 15-2. This adolescent girl with thyroid disease has irregular thickening of the pretibial skin (left panel). A skin biopsy shows a diffuse deposition of stromal mucin in the dermal interstitium (top right and first bottom right panels). It labels with the colloidal iron stain (second bottom right panel). |
With treatment of thyroid disease, pretibial myxedema has the potential to self-involute, with 70% of mild cases resolving after 25 years.3
Histologic Findings and Differential Diagnosis
One sees spaces between dermal collagen bundles, containing the mucinous deposits in the interstitium of the corium. This change may be subtle, especially in myxedema. The mucin is labeled with the colloidal iron or alcian blue techniques (Figure 15-3), and a diminution in the number of dermal elastic fibers may also be apparent with the Verhoeff-Van Gieson stain. There is no cellular proliferation in either myxedema or pretibial myxedema.
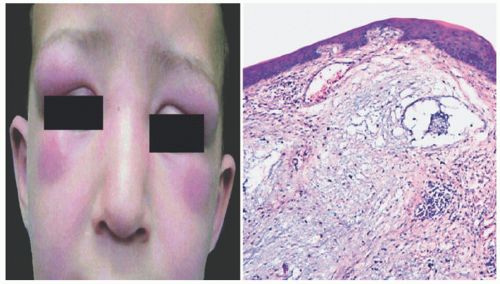 FIGURE 15-3. Self-healing mucinosis of the face in a young body, represented by periorbital swelling and erythema. Histologically, this process features a diffuse deposition of stromal mucin in the dermis, with the formation of mucin “pools.” |
Diagnosis is usually made with an evidence of characteristic skin lesions, a history of Graves’ disease, and the presence of Graves’ ophthalmopathy. Serological testing and skin biopsy can aid in diagnosis.
CAPSULE SUMMARY
MYXEDEMA AND PRETIBIAL MYXEDEMA
Myxedema and pretibial myxedema are both rare in children. The former is associated with congenital hypothyroidism. Pretibial myxedema is a thyroid dermopathy characteristic of Graves’ disease. Histologically, one sees spaces between dermal collagen bundles, containing the mucinous deposits in the interstitium of the corium. This change may be subtle, especially in myxedema. The mucin is labeled with the colloidal iron or alcian blue techniques.
CUTANEOUS MUCINOSIS OF INFANCY
Definition, Epidemiology, and Etiology
Clinical Presentation and Pathogenesis
CMI is characterized by firm, skin-colored to opalescent papules in a grouped, linear, or generalized distribution. The most commonly affected sites are the upper extremities, thighs, and trunk. The disorder usually presents at birth or shortly after and there is a slight female predominance.4,6 There is no systemic involvement with localized forms unlike other variants that may have associated thyroid abnormalities or paraproteinemias.7 The disease often has a persistent and gradual progression without tendency for self-involution. The distribution and the young age of affected individuals may raise the possibility that such lesions are connective tissue nevi of the proteoglycan type (nevus mucinosus).
Histologic Findings
One typically sees mucin in the papillary dermis with no fibroblastic proliferation and scant dermal perivascular chronic inflammatory cells. However, rare cases have histologic features that are reminiscent of adult scleromyxedema of the lichen myxedematosus type.
CAPSULE SUMMARY
CUTANEOUS MUCINOSIS OF INFANCY
CMI is a rare localized variant of cutaneous mucinoses with less than 10 reported cases. CMI is characterized by firm, skin-colored to opalescent papules in a grouped, linear, or generalized distribution. Histologically, there is mucin in the papillary dermis with no fibroblastic proliferation, and scant dermal perivascular chronic inflammatory cells.
SELF-HEALING JUVENILE CUTANEOUS MUCINOSIS
Definition, Epidemiology, and Etiology
Self-healing juvenile cutaneous mucinosis (SHJCM) is a form of localized primary cutaneous mucinosis.8 SHJCM is a rare disorder with less than 10 reported cases in the literature.9 The most common age of onset is between 5 and 15, although one reported case occurred in an infant less than 14 months of age.8 The etiology is unclear, but the proposed mechanism involves fibroblast stimulation by nonspecific antigens, including reports of SHJCM developing after viral illness, Bartonella exposure, and chemotherapy use in a nephroblastoma patient.9,10,11. There is no proven link between SHJCM and autoimmune disease. However, there are two reported cases of high-serum aldolase levels, suggesting a potential association with juvenile dermatomyositis.12
Clinical Presentation and Pathogenesis
The disease is characterized by three cutaneous findings: periorbital edema, deep-seated nodules, and ivory white papules. They are located most commonly on the face and periarticular regions.10,13 The distinguishing features reported by Bonerandi include young age at onset, the distribution of cutaneous findings on the face, scalp, neck, and joints; the absence of systemic symptoms including thyroid disease and paraproteinemia; acute onset; and spontaneous resolution.8 There are often associated arthralgias, fevers, and leukocytosis during the rapid proliferation phase. In most patients, the disease will clear within 7 months, but some reports have persisted for greater than 2.5 years.10
Histologic Findings
Microscopically, it features interstitial deposits of dermal stromal mucin, as described earlier in connection with myxedema. Fibroblastic proliferation has been reported only rarely. Scant perivascular chronic inflammation may again be apparent (Figure 15-4). Self-healing mucinosis may also manifest with subcutaneous nodules of mucin that are separated by fibrous septae, with limited chronic inflammation. In some instances, the nodules contain stellate, rhabdoid, or ganglion-like mesenchymal cells as well, yielding a superficial morphologic resemblance to proliferative fasciitis.
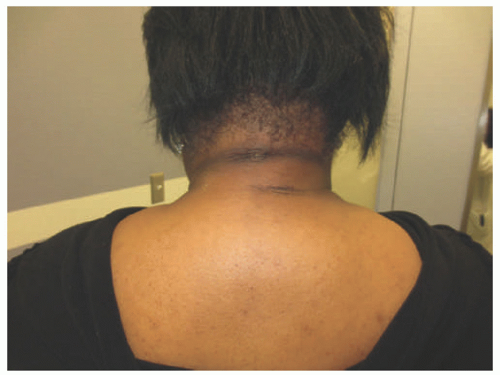 FIGURE 15-4. Scleredema of the mid upper back in a young African-American female. |
CAPSULE SUMMARY
SELF-HEALING JUVENILE CUTANEOUS MUCINOSIS
SHJCM is a rare disorder with less than 10 reported cases in the literature. The disease is characterized by three cutaneous findings: periorbital edema, deep-seated nodules, and ivory white papules. Microscopically, it features interstitial deposits of dermal stromal mucin, as described earlier in connection with myxedema.
SCLEREDEMA
Definition, Epidemiology, and Etiology
Scleredema, or Scleredema of Buschke, is a rare disorder characterized by progressive symmetric skin induration and thickening around the trunk, shoulders, and neck with associated woody nonpitting edema (Figure 15-5). There is often an association with Type I diabetes. There is no racial or ethnic predilection for any of the types, although there is a moderately increased propensity in females.14 Distal extremity involvement is very rare. Most cases occur before the age of 20.14,15,16,17
Clinical Presentation and Pathogenesis
There are three distinct entities. Type I is the most common occurring in the pediatric population, accounting for 55% of total cases. Type I usually has a preceding febrile illness, such as streptococcal pharyngitis, influenza, scarlet fever, or mumps and carries a good prognosis.17 The disease starts with an active inflammatory state with increased induration and skin thickening over 6 to 8 weeks followed by complete resolution and return to normal state by 6 months to 2 years.16 There are rare reports of progressively fatal disease.18 Type II accounts for 25% of cases and usually has an associated monoclonal gammopathy. Type III is slowly progressive and accounts for 20% of cases.
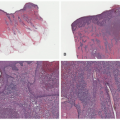
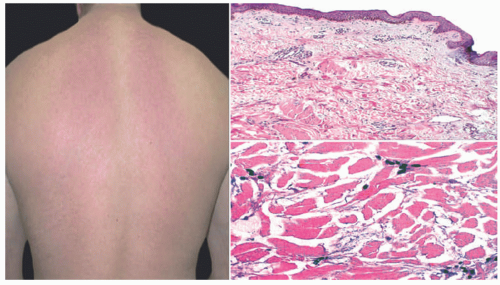 FIGURE 15-5. Scleredema in the skin of the back in an adolescent boy (left panel). Microscopically, this process is typified by a deposition of dermal interstitial stromal mucin, together with fibrosis (right panels). |
The pathogenesis is poorly understood. One proposed mechanism involves potential allergic sensitization by pathogens to collagen, leading to a disruptive antigenantibody reaction. Diabetes-associated cases may involve glucose-mediated fibroblast stimulation, leading to increased collagen production.15
Histologic Findings
The epidermis in scleredema may show partial effacement of the rete ridges and slight basilar hyperpigmentation. One regularly sees a thickened reticular dermis, with partial collagenization of the superficial subcutis. The collagen fibers are separated by variably dense deposits of interstitial mucopolysaccharide, and these tend to diminish as the duration of the disease increases. The mucin can be labeled with alcian blue or colloidal iron stains (Figure 15-4).
CAPSULE SUMMARY
SCLEREDEMA
Scleredema, or Scleredema of Buschke, is a rare disorder characterized by progressive symmetric skin induration and thickening around the trunk, shoulders, and neck with associated woody nonpitting edema. There is often an association with Type I diabetes. The epidermis in scleredema may show partial effacement of the rete ridges and slight basilar hyperpigmentation. The collagen fibers are separated by variably dense deposits of interstitial dermal mucin.
MUCOPOLYSACCHARIDE STORAGE DISEASES
Mucopolysaccharidoses (MPS) are a rare group of inherited lysosomal enzyme and storage deficiencies characterized by an aberrant catabolism of glycosaminoglycans. These fragments, including dermatan sulfate, heparin sulfate, and chondroitin sulfate, subsequently accumulate in various tissues. Affected children are often normal at birth and present with symptoms during infancy with a wide variation in phenotype.
Clinical Presentation and Pathogenesis
MPS can be classified into eight disorders, and the most common types will be discussed in the following paragraphs. All types of MPS can have many nonspecific cutaneous features, including prominent forehead, macroglossia, flat nasal bridge, and stubby hands.19,20,21
MPS I, or Hurler syndrome, is the most common type occurring in 1 of 100 000 births. Hurler syndrome is autosomal recessive with a deficiency in α-L-iduronidase, leading to the accumulation of heparin sulfate and dermatan sulfate. Infants present with widened nasal bridge and macroglossia by the first year of life, followed by progressive decline in cognition and motor skills with death from cardiorespiratory failure by age 10. They can also have generalized Mongolian spots, which can persist and progress into adulthood.20. Mongolian spots may be secondary to the association of heparin sulfate with high-affinity tyrosine kinase receptor and nerve growth factor, which are chemotropic for melanocytes.19
MPS II, or Hunter syndrome, is an X-linked recessive disorder with a deficiency in iduronate-2-sulfatase. The disease is rare with incidence 0.3 to 1.3 per 100 000 infants with male predominance. Children are often diagnosed later in childhood, around age 4 to 8. They can present with umbilical hernias and cardiomyopathy. They can also develop ivory papules in a reticular pattern over the scapula and lateral arms before age 10. These cutaneous findings are seen in 13% of patients and often have spontaneous regression.21 Death from systemic complications usually occurs around the third to fourth decade. Skin tightening and thickening is a feature of both MPS I and MPS II. This occurs most commonly on the hands and can lead to contractures, often mistaken for scleroderma.
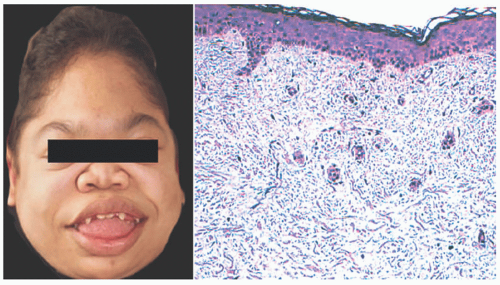 FIGURE 15-6. This female child has gargoylism, with distortion of the facial features caused by dermal deposition of mucopolysaccharide (left panel). Histologically, the dermis is largely effaced by mucoid material (right panel). |
Histologic Findings
Hurler and Hunter syndromes are typified pathologically by the presence of metachromatic granules in dermal fibroblasts, and occasionally in eccrine sweat glands as well. Interstitial mucin is also seen diffusely in the corium, and it can be highlighted with the Giemsa, alcian blue, and colloidal iron methods (Figure 15-6).
CAPSULE SUMMARY
MUCOPOLYSACCHARIDE STORAGE DISEASES
MPS are a rare group of inherited lysosomal enzyme and storage deficiencies characterized by an aberrant catabolism of glycosaminoglycans. All types of MPS can have many nonspecific cutaneous features, including prominent forehead, macroglossia, flat nasal bridge, and stubby hands. Hurler and Hunter syndromes are typified pathologically by the presence of metachromatic granules in dermal fibroblasts, and occasionally in eccrine sweat glands as well.
AMYLOIDOSES
Definition, Epidemiology, and Etiology
Amyloidosis can be characterized as localized, systemic, or familial. Primary localized cutaneous amyloidosis is a rare condition characterized by the deposition of extracellular
amyloid fibril deposition in the dermis without any systemic involvement. There are several different subtypes, including lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.22,23,24,25,26,27,28,29,30,31 Lichen and macular amyloidosis consist of keratin deposition, whereas nodular amyloidosis involves AL-type light chain deposition. Macular amyloidosis is characterized by aggregated brown papules with a rippled appearance, most commonly on the upper back. There is a strong female predominance, and it occurs most commonly in middle age.23 Nodular amyloidosis affects equal sexes and consists of waxy translucent nodules on acral sites, the face, and mucocutaneous junctions.23,24 There is a 9% risk of recurrence with nodular amyloidosis with a possible association with Sjogren syndrome.22,24
amyloid fibril deposition in the dermis without any systemic involvement. There are several different subtypes, including lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.22,23,24,25,26,27,28,29,30,31 Lichen and macular amyloidosis consist of keratin deposition, whereas nodular amyloidosis involves AL-type light chain deposition. Macular amyloidosis is characterized by aggregated brown papules with a rippled appearance, most commonly on the upper back. There is a strong female predominance, and it occurs most commonly in middle age.23 Nodular amyloidosis affects equal sexes and consists of waxy translucent nodules on acral sites, the face, and mucocutaneous junctions.23,24 There is a 9% risk of recurrence with nodular amyloidosis with a possible association with Sjogren syndrome.22,24
Lichen amyloidosis is a form of localized cutaneous amyloidosis characterized by intensely pruritic, hyperpigmented, discrete papules and plaques without any evidence of systemic involvement. This form of amyloidosis makes up 10% of all cutaneous amyloidoses.26 The disease occurs more commonly on the shins but has also been reported in the interscapular region and upper back.27 Although the disease is more common in the 5th and 6th decades of life, lichen amyloidosis has been reported infrequently in adolescents. 28 There is a slight male predominance in the South American, Chinese, and Southeast Asian populations.29 Lichen amyloidosis has been associated with atopic dermatitis, mycosis fungoides, systemic lupus erythematous, and multiple endocrine neoplasia (MEN) 2A or Sipple syndrome.30 Up to 36% of MEN 2A patients have this form of localized cutaneous amyloidosis.27 Etiology is unknown but may be related to viral and genetic factors, atopy, and localized friction.26 Pathogenesis is poorly understood but involves faulty neurotransmitter release in localized areas with resultant chronic rubbing and trauma. Repeated friction progresses to keratinocyte apoptosis and peptide degeneration. Dermal fibroblasts and macrophages convert these peptides into amyloid fibrils.29 The intense pruritus characteristically improves with sun exposure and worsens with stress.27 This process dictates clinical morphology with transition from intense pruritus to subtle hyperpigmentation to the formation of aggregated papules.31 Familial variants with autosomal dominant transmission have also been reported.31
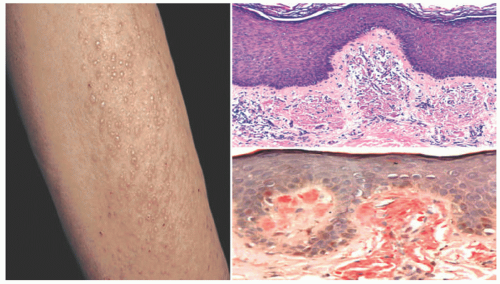 FIGURE 15-7. Lichen amyloidosis on the forearm of an adolescent boy, represented by small papulonodular lesions that were intensely pruritic (left panel). Microscopically, amorphous eosinophilic deposits are present in the upper dermis (top right panel), which label with the Congo red stain (bottom right panel). |
Familial amyloid deposition disorders include Muckle-Wells (urticarial-deafness-amyloidosis) syndrome, Familial Mediterranean Fever, and Schnitzler syndrome.23 Secondary cutaneous amyloidosis is usually the result of an underlying inflammatory process. Secondary forms can be seen in conditions such as basal cell carcinoma, porokeratoses, and adnexal tumors.25 The etiology involves aberrant keratinocyte apoptosis with filamentous degradation and deposition of amyloid.23 All forms of amyloidosis are extremely rare in the pediatric population.
Histologic Findings and Differential Diagnosis
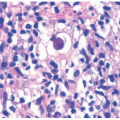
Both macular and lichen amyloidosis show amorphous eosinophilic deposits of amyloid in the dermal papillae (Figure 15-7). Apoptotic keratinocytes are present in the epidermis over the amyloid, often with pigment incontinence as well. The lesions of lichen amyloidosis may also include hyperkeratosis and acanthosis, as seen in lichen simplex chronicus. Cutaneous abnormalities in systemic amyloidosis likewise include dermal amyloid deposition, with virtually no inflammation. Amyloid is particularly seen in blood vessel walls, the basement membrane of sweat gland units, and adipocytes (Figure 15-8). In our experience, skin biopsies are as effective as fat-pad aspiration for the diagnosis of systemic amyloidosis.
Histochemical stains for amyloid include the Congo red, pagoda red, crystal violet, and thioflavine-T methods. It should be noted that affinity for Congo red is usually, but not always, absent in secondary systemic amyloidosis.
Immunohistochemical reagents are available for putatively specific staining of amyloid types AA and AL, but our experience with them has been disappointing diagnostically.
Immunohistochemical reagents are available for putatively specific staining of amyloid types AA and AL, but our experience with them has been disappointing diagnostically.
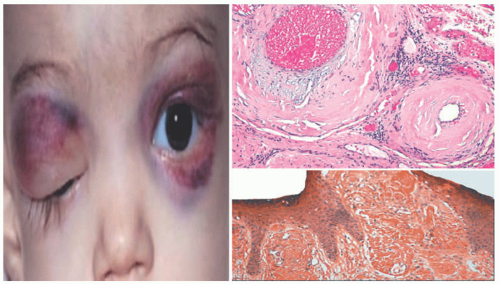 FIGURE 15-8. A child with systemic amyloidosis has “raccoon eyes,” which are the result of vascular fragility in periorbital blood vessels (left panel). Microscopically, amorphous eosinophilic deposits are present in the walls of deep dermal blood vessels (top right panel), and these label with the Congo red stain (bottom right panel). |
Cutaneous amyloid deposits may resemble the material seen in juvenile colloid milia. However, the latter do not label with Congo red and instead are immunoreactive for keratin. Other histologic differential diagnostic considerations include erythropoietic protoporphyria and lipoid proteinosis, in which acellular eosinophilic intradermal material may also be present. The eosinophilic material does not label with histochemical stains for amyloid in either of those conditions.
CAPSULE SUMMARY
AMYLOIDOSES
Amyloidosis can be characterized as localized, systemic, or familial. Primary localized cutaneous amyloidosis is a rare condition characterized by the deposition of extracellular amyloid fibril deposition in the dermis without any systemic involvement. There are several different subtypes, including lichen amyloidosis, macular amyloidosis, and nodular amyloidosis. Familial amyloid deposition disorders include Muckle-Wells (urticarial-deafness-amyloidosis) syndrome, Familial Mediterranean Fever, and Schnitzler syndrome. Both macular and lichen amyloidosis show amorphous eosinophilic deposits of amyloid in the dermal papillae.
CALCINOSIS CUTIS
Definition, Epidemiology, and Etiology
Calcinosis cutis is a rare disorder characterized by the deposition of calcium phosphate or hydroxyapatite in areas of microtrauma. Most cases are diagnosed before the age of 10, approximately 2 to 3 years after diagnosis. A possible etiology involves the release of calcium from muscle cells affected by myopathy.32,33
Clinical Presentation and Pathogenesis
The deposition of calcium phosphate or hydroxyapatite leads to the formation of palpable yellow to white nodules with or without associated contractures (Figure 15-9). Local inflammation can subsequently lead to ulceration or the discharge of milky white substrate. There is predilection for the forearms, elbows, and fingers.32,33,34,35,36
Calcinosis cutis can be classified into four subtypes: metastatic, dystrophic, idiopathic, and iatrogenic.34,35 Metastatic calcinosis cutis occurs with elevated serum calcium and or phosphate levels and can be seen in conditions such as sarcoidosis, chronic renal failure, or hyperparathyroidism.
Dystrophic calcinosis is the most common subtype. It occurs in previously inflamed tissues in the setting of normal serum calcium and phosphate levels. Dystrophic calcinosis can be seen with CREST syndrome, system lupus erythematosus, and approximately 30% to 70% of juvenile dermatomyositis. This form is also seen in inherited syndromes such as Ehler-Danlos, Werner, and pseudoxanthoma elasticum.32,33,36 Ulceration and infection are common in juvenile dermatomyositis, and calcinosis can be superficial or extend to the underlying fascia and muscle with limitations in movement.32
Dystrophic calcinosis is the most common subtype. It occurs in previously inflamed tissues in the setting of normal serum calcium and phosphate levels. Dystrophic calcinosis can be seen with CREST syndrome, system lupus erythematosus, and approximately 30% to 70% of juvenile dermatomyositis. This form is also seen in inherited syndromes such as Ehler-Danlos, Werner, and pseudoxanthoma elasticum.32,33,36 Ulceration and infection are common in juvenile dermatomyositis, and calcinosis can be superficial or extend to the underlying fascia and muscle with limitations in movement.32
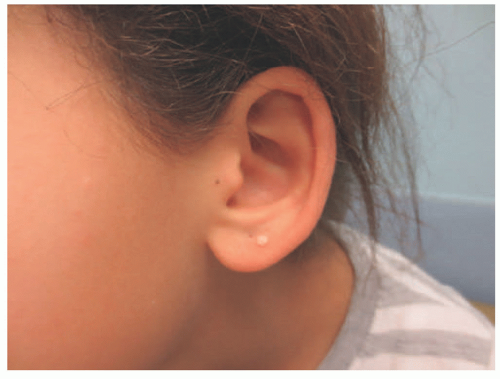 FIGURE 15-9. Calcinosis cutis of the lobule in a young healthy girl without any underlying systemic illness. |
Idiopathic calcinosis is a diagnosis of exclusion that also occurs in the setting of normal serum calcium and phosphate levels. It can often be seen with Down syndrome and scrotal calcinosis (Figure 15-10). Iatrogenic calcinosis cutis is often seen after the placement of calcium electrodes for electrocardiograms or repeated heel pricks in neonatal infants in the intensive care unit. Heel stick calcinosis appears usually by 4 months of age with spontaneous resolution by 18 to 30 months of age. The etiology of this condition involves repeated phlebitis with the release of alkaline phosphatase and increased pH, leading to calcium salt deposition.35 A second pathogenesis has been described, which involves tissue damage with denatured proteins that preferentially bind phosphate. Calcium then binds phosphate ions, leading to precipitation.34
Histologic Findings
All forms of calcinosis cutis feature the histologic presence of calcium phosphate deposits in the dermis or subcutis, unassociated with inflammation or foreign body reaction (Figure 15-10). The lesions can be labeled with histochemical stains for calcium, such as the von Kossa or alizarin-red methods.
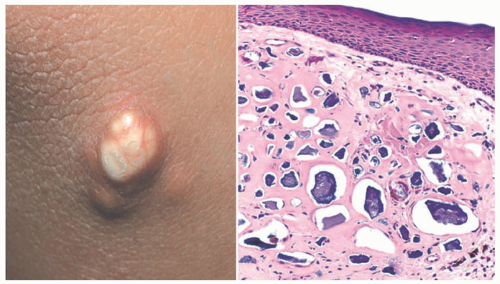 FIGURE 15-10. Calcinosis cutis on the buttock in a child, taking the form of hard white nodular lesions (left panel). Histologically, basophilic calcium salts are distributed throughout a fibrotic dermis (right panel). |
CAPSULE SUMMARY
CALCINOSIS CUTIS
Calcinosis cutis is a rare disorder characterized by the deposition of calcium phosphate or hydroxyapatite in areas of microtrauma. The deposition of calcium phosphate or hydroxyapatite leads to the formation of palpable yellow to white nodules with or without associated contractures. All forms of calcinosis cutis feature the histologic presence of calcium phosphate deposits in the dermis or subcutis.
OSTEOMA CUTIS
Definition, Epidemiology, and Etiology
Osteoma cutis is characterized by heterotopic bone formation in the skin and consists of firm, nontender subcutaneous papules and nodules. It is further classified as primary or secondary, with the latter applying if the process arises in a preexisting skin lesion.37 Primary osteoma cutis is associated mostly with inherited syndromes with inactivating mutations in GNAS including Albright’s Hereditary Osteodystrophy (AHO), progressive osseous heteroplasia, fibrodysplasia ossificans progressiva, or plate-like osteoma cutis.38,39,40 Osteoma cutis is seen in 25% to 50% of patients with AHO and presents in early infancy.41
Clinical Presentation and Pathogenesis
Plate-like osteoma cutis presents in newborns and young children with large skin-colored plaques ranging from 1 to 15 cm on the scalp38 (Figure 15-11). Progressive osseous heteroplasia is the most severe form, presenting with dermal ossification in infancy with extension to skeletal
muscle.37 Secondary osteoma cutis accounts for 85% of total cases. This subtype arises from preexisting skin lesions, the sites of prior trauma, or surgical procedures including scars, acne, and basal cell carcinoma. Miliary osteoma cutis usually occurs in females at the sites of prior acne scars.38,42
muscle.37 Secondary osteoma cutis accounts for 85% of total cases. This subtype arises from preexisting skin lesions, the sites of prior trauma, or surgical procedures including scars, acne, and basal cell carcinoma. Miliary osteoma cutis usually occurs in females at the sites of prior acne scars.38,42
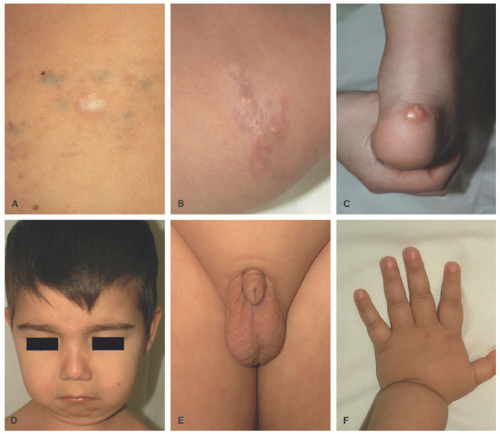 FIGURE 15-11. Osteoma cutis in the setting of Albright hereditary osteodystrophy. Osteoma cutis: a bluish or erythematous, indurated plaque on the back (A) and the lower limb (B), or a reddish, stone-hard, dome-shaped nodule located on the heel (C). D, A characteristic round face with flattened nasal bridge and short neck. E, Micropenis. F, A typical short, stubby hand with brachydactyly. Reprinted with permission from Kacerovska D, Nemcova J, Pomahacova R, Michal M, Kazakov D. Cutaneous and superficial soft tissue lesions associated with albright hereditary osteodystrophy: clinicopathological and molecular genetic study of 4 cases, including a novel mutation of the GNAS gene. Am J Dermatopathol. 2008;30(5):417-424. |
Pathogenesis is unknown, but it is speculated to involve differentiation of mesenchymal cells, such as fibroblasts, into osteoblasts.43 Prognosis is good, with most cases remaining stable. However, patients should be monitored for associated conditions and progression to cosmetic defects.37
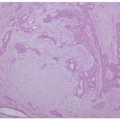
The mechanism of heterotopic bone formation in the skin is not altogether clear, but, as stated earlier, one theory suggests that fibroblasts in the dermis undergo metaplasia to osteoblasts, as a result of abnormalities in the genes that govern bone formation.44 A skin biopsy is necessary for a definitive diagnosis of osteoma cutis, in which mature cortical bone is present in the dermis or subcutis (Figures 15-12 and 15-13). Conventional nevus with secondary ossification (osteonevus of Nanta) is seldom encountered in routine dermatopathologic practice. This type of nevus is usually located in the upper part of the body, with the predilection in females.
Histologic Findings
The ossification is usually in the form of the small islands of compact lamellar or structureless bone at the base of the lesion (Figure 15-14). In some cases, trabecular bone with bone marrow and adipocytes may also be present.
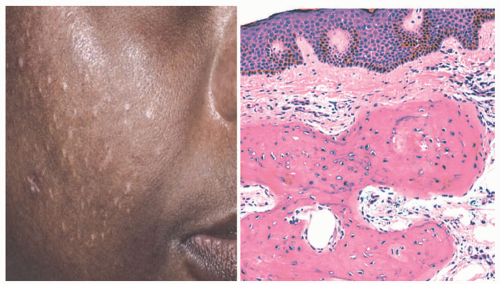 FIGURE 15-12. An adolescent girl had scarring acne vulgaris and has now developed firm white papules in the facial skin (left panel). Microscopically, they represent osteomas that are centered on the former foci of acne (right panel). |
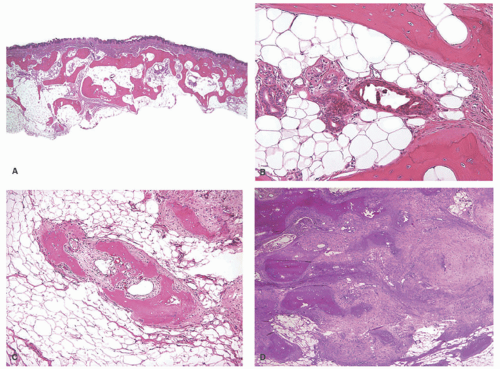 FIGURE 15-13. The so-called plaque-like osteoma in the setting of Albright hereditary osteodystrophy. The bone grows in a horizontal plaque-like fashion (A), often wrapping around eccrine or apocrine ducts, some of which manifested dilatation and hyperplasia with intraluminal bridges (B). Although the bone lamellae are sharply segregated from the surrounding fat (C), another lesion from this patient showed curved bone in the background of immature mesenchymal cell proliferation, occasioning resemblance to fibrous dysplasia (D). Reprinted with permission from Kacerovska D, Nemcova J, Pomahacova R, Michal M, Kazakov DV. Cutaneous and superficial soft tissue lesions associated with albright hereditary osteodystrophy: clinicopathological and molecular genetic study of 4 cases, including a novel mutation of the GNAS gene. Am J Dermatopathol. 2008;30(5):417-424. |
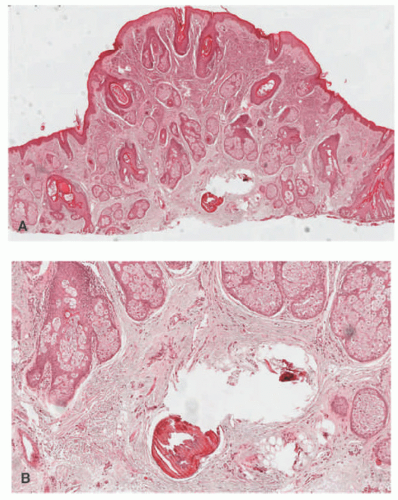 FIGURE 15-14. Osteonevus of Nanta. A, There is an intradermal melanocytic nevus with a congenital pattern (nested melanocytes are tracking down adnexal structures). B, At the base of the lesion, a focus of metaplastic bone formation is present. Digital slides courtesy of Path Presenter.com. |
CAPSULE SUMMARY
OSTEOMA CUTIS
Osteoma cutis is characterized by heterotopic bone formation in the skin and consists of firm, nontender subcutaneous papules and nodules. The ossification is usually in the form of the small islands of compact lamellar or structureless bone at the base of the lesion.
ARGYRIA
Definition, Epidemiology, and Etiology
Argyria is a rare condition characterized by the development of slate gray to blue mucocutaneous discoloration following chronic exposure to silver-containing products. The discoloration is likely secondary to melanocyte stimulation and degradation of elemental silver.45 Argyria is worsened by sunlight, which can act as a catalyst for the reduction of silver. The dyspigmentation can take several months to appear after the chronic use of silver-containing products.31 Several culprits have been reported in the literature, including the use of colloidal silver supplements in cystic fibrosis patients for mucous clearance and topical silvadene cream in a young adult with dystrophic epidermolysis bullosa.46,47 Despite the discontinuation of silver-containing products, the discoloration can persist lifelong.
Histologic Findings
Microscopically, silver granules in argyria are found along the basement membranes of sweat glands, hair follicles, and the epidermis. They are most easily seen as refractile granules with darkfield microscopy (Figures 15-15 and 15-16). Slight melanosis of the epidermis or dermal melanin incontinence may accompany the silver deposition.
CAPSULE SUMMARY
ARGYRIA
Argyria is a rare condition characterized by the development of slate gray to blue mucocutaneous discoloration following chronic exposure to silver-containing products. Microscopically, silver granules in argyria are found along the basement membranes of sweat glands, hair follicles, and the epidermis.
Cutaneous Cysts
Cutaneous cysts are among the most commonly seen lesions in dermatopathology, and they are seen in patients of all ages. Simply defined, a “cyst” is an enclosed space in a tissue compartment that is lined by epithelium and that contains fluid or semisolid material. Most cysts in the skin are thought to derive from the dermal appendages, as “retention” phenomena. Developmental cysts are less common, having their origins in the vestigial remnants of embryonic tissue.
As one might expect, the epithelial lining of cutaneous cysts is potentially variable in nature morphologically, reflecting the spectrum of differentiation that is seen in the normal epidermis and its appendages. Salient pathologic features of the lesions discussed in this chapter are presented in Table 15-1. Additional types of cysts exist in the skin, but they are seen in adults and therefore have been omitted from consideration here.
TRUE CUTANEOUS CYSTS
Epithelial Cysts Lacking Ciliated Cells
EPIDERMAL (EPIDERMOID) CYST, MILIA, AND COMEDONAL CYST
Definition, Epidemiology, Clinical Presentation, and Pathogenesis
Epidermal, or infundibular cysts are flesh-colored firm mobile papules and nodules, often with central puncta (Figure 15-17). Rupture may occur either spontaneously or
as a result of trauma, with the extrusion of foul-smelling contents and secondary inflammation. This incites an inflammatory foreign body-type reaction to keratinous cyst contents in the surrounding tissue. They are twice as common in males as in females and usually appear in the third to fourth decade of life but can arise during childhood or puberty.48 In the pediatric population, approximately 13% of all head cysts are characterized as epidermoid cysts and favor the eyebrows and postauricular region. Epidermoid cysts are also associated with several genetic syndromes, including Gardner syndrome, occurring in 36% to 50% of these patients.49 Gardner syndrome is an autosomal dominant disease complex caused by mutations in the APC gene. Affected individuals may have multiple adenomatous
intestinal polyps, soft tissue fibromas and desmoid tumors, osteomas, and cutaneous cysts beginning in childhood. The risk for the eventual development of colorectal adenocarcinoma is high.
as a result of trauma, with the extrusion of foul-smelling contents and secondary inflammation. This incites an inflammatory foreign body-type reaction to keratinous cyst contents in the surrounding tissue. They are twice as common in males as in females and usually appear in the third to fourth decade of life but can arise during childhood or puberty.48 In the pediatric population, approximately 13% of all head cysts are characterized as epidermoid cysts and favor the eyebrows and postauricular region. Epidermoid cysts are also associated with several genetic syndromes, including Gardner syndrome, occurring in 36% to 50% of these patients.49 Gardner syndrome is an autosomal dominant disease complex caused by mutations in the APC gene. Affected individuals may have multiple adenomatous
intestinal polyps, soft tissue fibromas and desmoid tumors, osteomas, and cutaneous cysts beginning in childhood. The risk for the eventual development of colorectal adenocarcinoma is high.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


