31 Vascular anomalies of the upper extremity
Synopsis







Introduction
During the past four decades, there has been a steady, almost exponential increase in our knowledge of vascular anomalies. A biological classification system has evolved and is under constant refinement.1 A careful physical examination augmented with advanced imaging will yield an accurate diagnosis from the vast myriad of possibilities. A multidisciplinary team can offer individualized treatment with some degree of predictability. In less than 40% of these lesions, one option is surgery, which is fraught with potential complications and poor outcomes. However, in many patients surgical treatment is often the best remedy and should not be viewed as the last resort. A carefully planned and well-orchestrated procedure and rehabilitation can yield remarkable outcomes.2
History and classification
The etiology of vascular birthmarks has long stimulated the fertile imagination of man. At any point in time, classification systems have reflected a balance between folklore and science. Well into the 19th century, the doctrine of maternal impressions dictated that a gravid mother’s craving for strawberries, sight of an accident, or emotional longing could imprint a vascular blemish, a naevus maternus, on her unborn child.3 With the development of the microscope a vast number of histological terms have been introduced and to this day, most of these lesions are inaccurately labeled as hemangiomas. Study of the histopathology and natural history of these lesions spurned intriguing embryological classifications,4,5 which seemed logical at the time but failed to distinguish involuting from noninvoluting lesions and offered little guidance in management. In the late 1970s, Mulliken and Folkman proposed a working hypothesis that these cells could be distinguished by their cellular characteristics and subsequently surgical specimens were analyzed by selected histological stains, electron microscopy and autoradiography (uptake of tritiated thymidine into DNA). On the basis of these studies two types of lesions were present: those, which showed endothelial hyperplasia designated as hemangiomas, and those, which did not, called malformations. A binary schema based on biologic activity was proposed1 and accepted by the International Society for the Study of Vascular Anomalies (ISSVA) in 1996.6 This system has been corroborated by radiological studies7,8 and immunohistological staining.9 However, “no system is carved in stone and must be written on recyclable paper” and is in a constant state of revision. For example, the initial category of hemangiomas has been changed to vascular tumors to include all vascular tumors in all age groups. A particular vascular lesion may be moved from one category to another or may be discarded completely with our expansion of clinical experience and basic science investigation. For the past 30 years, this system has been very helpful to the upper limb surgeon in the proper diagnosis and management of these lesions (Table 31.1).2 Most frustrating are the large number of eponyms with and without abbreviations, which have been attached to these lesions. These terms are often used indiscriminately without specific reference to cell type, flow characteristics and growth. Adding to the confusion is the common association of growth disturbances. Both overgrowth and undergrowth with and without dysplastic fat deposition may accompany vascular anomalies. The summary in Table 31.2 helps delineate our present state of knowledge of this nosologic quagmire.
Table 31.1 ISSVA classification of vascular anomalies
| Vascular tumors | Vascular malformations |
|---|---|
| Slow-flow vascular malformations | |
| Hemangioma | Capillary malformations CM) |
| Infantile hemangioma (IH) | Port-wine stain |
| Congenital (CH) | Telangiectasias |
| Noninvoluting congenital hemangioma (NICH) | Angiokeratoma |
| Rapidly involuting congenital hemangioma (RICH) | Venous malformations (VM) |
| Hemangiomatosis | Common sporadic VM (VM) |
| Pyogenic granuloma | Blue rubber bleb syndrome (BRBS) or Bean syndrome |
| Kaposiform hemangioendothelioma KHE) | Glomuvenous malformation (GVM) |
| Rare tumors: | Familial cutaneous and mucosal venous malformation (VMCM) |
| Hemangioendothelioma | Maffucci syndrome |
| Infantile fibrosarcoma | Diffuse VM (Bockenheimer) |
| Hemangiopericytoma | Lymphatic malformations (LM) |
| Giant cell angioblastoma | Common lymphatic malformation (LM) |
| Lymphangiomatosis | |
| Gorham-Stout syndrome | |
| Fast-flow vascular malformations | |
| Arterial malformation (AM) | |
| Arteriovenous malformation (AVM) | |
| Arteriovenous fistula (AVF) | |
| Complex-combined vascular malformations | |
| CVM, CLM, LVM, CLVM (Klippel–Trenaunay), | |
| CAVM (Parkes–Weber), AVM-LM,CM-AVM | |
| Proteus, hemihypertrophy, capillary-lymphatic-overgrowth-vascular-epidermal nevus (CLOVE) | |
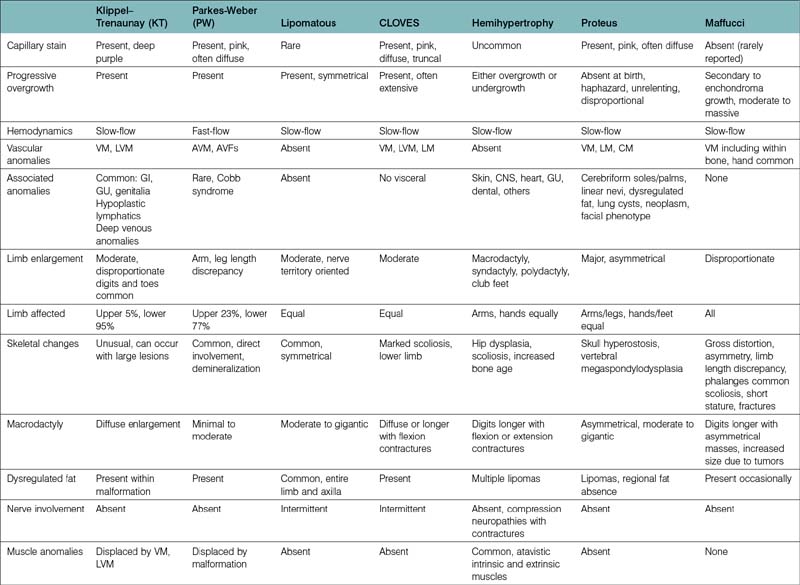
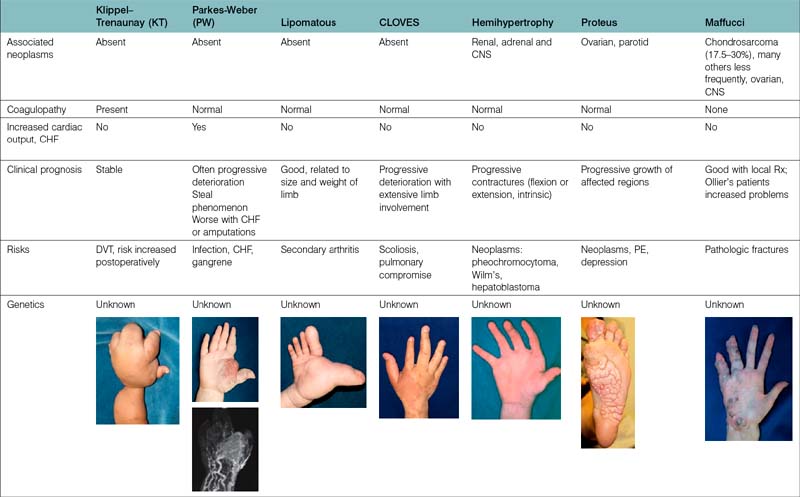
A correct diagnosis impacts directly upon appropriate treatment and an incorrect diagnosis may result in unnecessary or harmful remedies. Most upper limb surgeons designate all fast flow anomalies with or without arteriovenous fistulae as Parkes–Weber syndrome (PWS). However, PWS patients comprise a very small percentage of the fast flow group, which comprises about 15% of upper extremity fast-flow malformations.10 Not all slow flow combined malformations should be called Klippel–Trenaunay syndrome (KTS), an eponym we reserve for combined CLVM lesions. The VMs collectively comprise the largest clinical group of patients evaluated by the hand surgeon. The majority are small or large lesions involving all portions of the upper limb and/or ipsilateral chest wall. Other subgroups have emerged. The diffuse VMs involving all structures within the upper limb including bone have been designated Bockenheimer lesions,11 originally described as “diffuse genuine phlebectasias”, and are not amenable to surgery with the exception of pathologic fractures and localized areas of symptomatic thromboses. Maffucci patients are commonly associated with skeletal enchondromas but many of the skeletal lucencies may be clusters of VMs, which are also found in abundance within the soft tissue planes.12 The glomuvenous (GVM) lesions occur in clusters with a characteristic appearance, are painful, have been localized to chromosome 1p21–22, and in the past were called glomus tumors or glomangiomas.13 They have been renamed to stress the fact that they are malformations and not tumors as suggested by the suffix “-oma.” The VMs seen in the blue rubber bleb nevus syndrome (BRBN) also have a characteristic cutaneous appearance, are also familial and often confused with GVM, Maffucci and CM-VM.14 The associated gastrointestinal lesions, often localized to the small bowel, are often the cause of anemia and iron deficiencies (see Table 31.7).14,15
Lymphatic malformations often occur in combination with other vascular elements but the most common varieties are either isolated or diffuse LMs, which may involve any portion of the upper limb and/or ipsilateral chest wall.10 Lymphangiomatosis conditions are not of primary concern to the upper limb surgeon as they involve visceral organs and/or the lungs and in the long run are life endangering. LMs in the Gorham–Stout syndrome involve skeletal structure including the humerus, radius, ulna and tubular bones of the hand.16 Pathologic fractures can occur.
There are many conditions in which vascular anomalies, overgrowth or undergrowth co-exist and with our present state of knowledge, it is difficult to draw clear lines due to the increasing amounts of growth factors found in the omnipresent dysplastic fat tissue within these extremities. Often at their vascular clinic reviews, the authors encounter lesions that do not fit into any specific category and are designated as “PUVA”, provisionally unique vascular anomalies. With time, this large group of “unknowns” will become more precisely defined. The emergence of the CLOVES syndrome is a good example, important to the hand surgeon. Children with this present with condition large extremity and truncal CMs, moderate to marked overgrowth, large fat accumulation within all tissue planes, either LMs or VMs and epidermal nevi.17,18 Previously, most had been called “macrodactylies”, “gigantism” or Proteus and did not fit the criteria for the Proteus syndrome (Table 31.2).19
Another group of confusing lesions can be found in the fast-flow group where all lesions should not be called Parkes-Weber anomalies. Most fast-flow lesions are easily diagnosed by their clinical characteristics, palpable bruits, shunts and progressive growth. A small group of these children seen by the extremity surgeon may have mild hypotonia, lipomas in any subcutaneous location, fast flow deeper lesions and frontal bossing. Males have a nevus on the glans of the penis. In the past they were placed in the Bannayan-Riley-Ruvalcaba (BRR) syndrome or the Cowdan syndrome. Genetic testing is diagnostic and they are now called the PTEN hamartoma-tumor syndrome (PHTS) (see Table 31.8).20,21 The hand surgeon may also see the capillary malformation-arteriovenous malformation (CM-AVM) syndrome, which initially appears to be an innocent CM, which are pinkish red and surrounded by a small halo. One-third of these patients may have an existing AVM and 12% may evolve into a Parkes–Weber syndrome (PWS) and intracranial AVMs should be considered. This condition is hereditary and results in a mutation in RASA1.22 The PWS describes a diffuse AVM of an overgrown extremity with an overlying CM.2,23,24 In contrast to lesions with direct arterio-to-venous shunting (AVMs with AVFs), PWS lesions show diffuse involvement of all soft tissue structures with fast flow arterioles in the initial absence of large draining veins. Large shunts develop secondarily. The lower extremity is involved much more frequently than the upper.
Diagnosis/patient presentation
Evaluation begins with a detailed history and physical examination. In contrast to the rapid growth and slow involution that characterize infantile hemangioma (IH), vascular malformations expand commensurately with the child. Although vascular malformations are by definition present at birth, many are inconspicuous and some do not appear until late childhood. Dermal ectasias, such as the relatively common capillary malformation (“port-wine stain”), are usually noted in the newborn nursery. Rarely, later in life will it become obvious that some of these capillary stains overlie a slow-flow (venous or lymphatic) anomaly or a fast-flow (arteriovenous) malformation. Venous and lymphatic malformations often co-exist and may be quite insidious in their initial presentation. Venous malformations may not manifest until late childhood or adolescence; large lesions are usually obvious within the first 4–8 years of life. Fortunately, the vast majority of vascular malformations are correctly diagnosed by clinical examination alone.3,25,26 It is important to obtain baseline functional data including length and circumference measurement of the lower limbs early in life and to follow these children as they grow. The clinical characteristics of the more common malformations will be described with the individual case presentations.
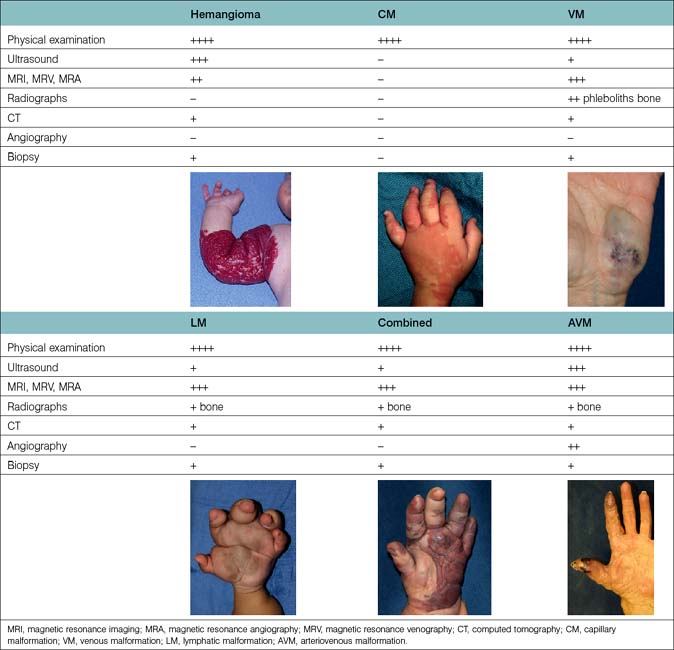
The diagnostic work-up varies for each type of vascular anomaly (Table 31.3). Ultrasonography, sometimes with Doppler scans, confirms the initial distinction between a vascular tumor and vascular malformation.27 These studies are easily performed in the clinic. This diagnostic tool not only pinpoints the location of the lesion but also enables its identification as either slow-flow or fast-flow. Expensive diagnostic studies are unnecessary in the case of a diffuse or localized, asymptomatic slow-flow malformation or a small fast-flow lesion amenable to simple excision and closure. When a patient is symptomatic or if an operation is being considered for a large vascular anomaly, one should learn as much as possible about the size, extent, depth, and three-dimensional characteristics and obtain specific information about the location, caliber, and flow characteristics. Magnetic resonance imaging (MRI) with contrast (gadolinium) is the “gold standard.” Fat suppressed T-1 weighted sequences with contrast will differentiate between a vascular tumor and malformation.28 Computed tomography (CT) can be used to detect interosseous involvement or skeletal distortion, caused by a vascular malformation, and help delineate soft tissue planes. Large cystic cavities in an LM are clearly depicted. New three-dimensional rendering of both fast-flow and slow-flow malformations using CT technology provides very impressive views of the entire malformation and its relationship to neighboring soft tissue and skeletal structures. Nevertheless, it is questionable whether CT angiography (CTA) is really useful in planning therapy. However, formatted three-dimensional rendering of CTA can be invaluable preoperatively. Plain radiology of the affected region provides very little information other than showing the presence of phleboliths and interosseous involvement or other skeletal distortions. Angiography has very little use in the diagnosis of slow-flow malformations, although it is indispensable in the diagnosis and assessment of fast-flow lesions.2,10 The angio-architecture and three-dimensional anatomy of the malformation can be precisely determined and the arterial suppliers and draining veins clearly identified. Indirect and direct phlebography techniques are sometimes used for diagnostic and therapeutic purposes for VM in the extremity.
Treatment/surgical technique
 Video1
Video1Surgical principles gain acceptance with time and must be periodically reassessed and refined, particularly in this rapidly advancing field. Principles should not be confused with surgical techniques that change frequently, and are modulated to a great extent by technology. Many of the following principles have evolved from the early operative experience with vascular malformations (especially the fast flow types), which was punctuated with complications (Table 31.4).2,3
1. Preoperative planning should include correlation of the size, extent, and involvement of structures with physical examination, MRI scans, radiographs, and angiograms. All studies should be reviewed preoperatively; serial studies of growing children are often invaluable in demonstrating the true extent of involvement of the extremity. A thorough explanation of all potential complications must be given to the patient and/or parents.
2. The surgeon should mentally outline the extent of the resection and abide by it. Use of the pneumatic tourniquet and complete exsanguination is necessary to visualize normal and abnormal structures clearly, regardless of the size of the malformation. Once the operative field becomes bloodstained, the identification of nerves, tendons, intrinsic muscles, and other structures is more difficult.
3. Placement of incisions is important – particularly in children. A high mid-axial incision in the digit is preferred, as it can be used again and is hidden. With growth, palmar scars within or near the palm can lead to contracture. If a palmar approach is chosen, a zig-zag incision is preferred, utilizing existing skin creases. If multiple debulking procedures are contemplated in the upper arm or forearm, each incision should be planned carefully to avoid unnecessary scarring. The medial surface of the arm, elbow and forearm is the least conspicuous. Incisions in the weight-bearing plantar surfaces of the foot should be avoided. The placement of incisions in the medial arch, along the borders of the foot, and on the dorsal surfaces is safe. Scars in the web spaces or along eponychial and/or paronychial folds will contract and are likely to become problematic.
4. Magnification, either loupe or microscopic, makes a tremendous difference in the identification and preservation of normal neurovascular structures. Small vessels, such as vincular pedicles to the flexor tendons and nutrient vessels to the carpal bones, should be saved if uninvolved. Often, vascular anomalies will displace, but not typically invade, neighboring soft tissues.
5. Thorough dissection limited to a specified area should be performed so that subsequent re-entry into a densely scarred bed will be unnecessary. Often, the malformation extends well beyond its anticipated limits. In certain regions, limited staged excisions are better than one extensive and protracted dissection that leaves behind tissue containing abnormal vasculature. For example, the digits (including the thumb) should be debulked in stages. A single procedure is usually best for the dorsum of the hand, wrist, and palmar surface of the same in a patient with a diffuse LM or VM.
6. Avoid vascular compromise. Only one-half of a digit should be dissected at a time (see Table 31.6). If possible, at least one or two large dorsal veins per finger should be preserved to enhance venous drainage, especially when excising a diffuse lymphatic lesion in the hand or foot. The superficial venous system should not be resected unless a deep system has been identified. When a critical arterial segment to a digit, hand or foot is removed, this segment should be reconstructed with a vein graft so that at least one digital artery is preserved per digit and one major artery supplies the hand or foot with a functional intact palmar or plantar arch.
7. Avoid intraneural dissection whenever possible, despite gross involvement. Although many vascular malformations, particularly the venous type, are entangled in nerves, dissection often leaves in-continuity neuromas with partial or complete loss of distal sensory or motor function. The symptoms are usually much worse than the initial ones.
8. Avoid partial dissections within large muscle groups. Removal of the entire muscle en bloc is preferred to avoid a secondary contracture of the entire musculotendinous unit. If more than half of a skeletal muscle is excised piecemeal, a secondary contracture is likely. VMs, mixed CLVMs, CAVMs and LVMs are the most problematic lesions. Pure LMs may extend beneath the muscular fascia but expand along fascial planes and usually do not penetrate the muscles themselves.
9. Cutaneous flaps should not be inset under tension and nonviable skin should be replaced with a skin graft or a flap. Tissue with questionable viability can be observed and resected later, if necessary. It is usually best to remove and replace skin that is heavily scarred from a previous procedure, chronically infected and ulcerated, populated by coalesced lymphatic vesicles or deprived by a proximal steal phenomenon.
10. Drains should be used liberally, and delayed primary closure of the wound should be considered. Persistent postoperative bleeding is usually best treated with direct pressure, elevation, and immobilization, rather than by re-exploration. Baseline coagulation studies should be performed for large/diffuse VM and LVM. Liberal use of tissue sealant products is encouraged.
11. Immobilize the operated extremity in a young child or adolescent. In the treatment of children with vascular anomalies, the postoperative lack or loss of immobilization is the single most important cause of wound dehiscence, maceration, and chronic infection.
12. The surgeon who has the courage to treat difficult fast-flow lesions should also be prepared to amputate a symptomatic nonfunctional or painful digit, hand, leg or foot following unsuccessful attempts at palliation. The reconstructive surgeon need not view amputation as a failure.
13. Follow-up evaluation should be performed compulsively at yearly intervals, regardless of the particular type of malformation, size, and hemodynamic activity. The true extent of the lesion can never be absolutely eradicated short of an amputation. Early childhood, adolescence, and pregnancy are times when change may occur in vascular malformations due to hormonal influences. These patients and parents often have questions that are not easily answered by their family physician owing to the paucity of information available in the medical literature. Both slow-and fast-flow lesions may expand dramatically during pregnancy, with use of high estrogen anti-ovulation medication, or following trauma. Large AVMs, LMs or mixed lesions may be well tolerated by young children only to become burdensome to teenagers due to their expansion, bulk, ulceration, and appearance or steal symptoms.
14. Give the patient and parents adequate time to make their decision. The diagnosis and treatment of vascular anomalies is a rapidly changing field. It is important to keep both parents and patients at the appropriate age clearly informed about the natural history of their particular malformation, options for treatment and new types of therapies. It is axiomatic that the surgeon provide a clear explanation of all potential complications and expected short and long term outcomes prior to any surgical treatment.
Table 31.4 Principles of management
Caveats of palmar dissections. Multiple technical refinements have made these dissections, once considered impossible, both predictable and safe. These are summarized in Table 31.5.
Table 31.5 Caveats of palmar dissections
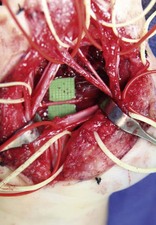
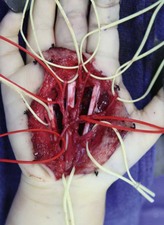
Caveats of thumb/digital dissections. Surgical approaches to the thumb and digit are similar to those summarized above and have become much safer with microvascular techniques. These are summarized in Table 31.6.
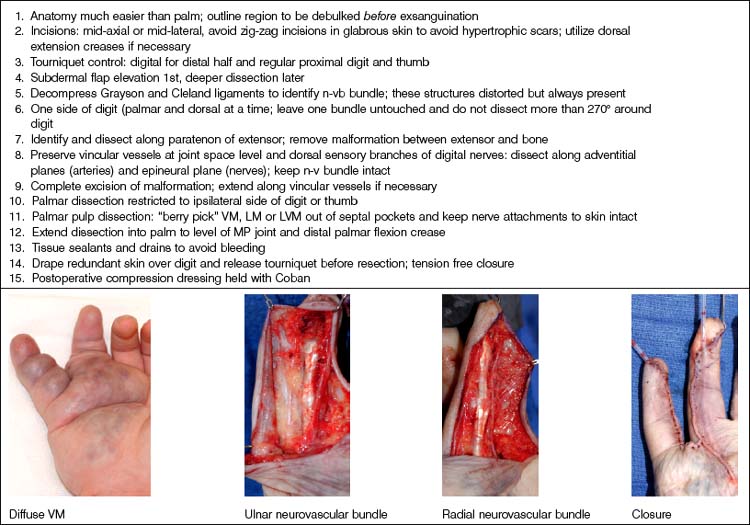
Vascular tumors
Infantile hemangioma (IH)
Basic science/disease process
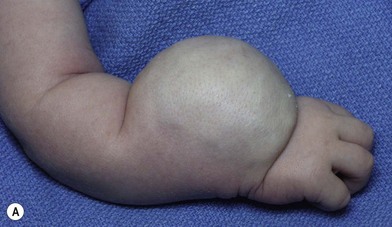
IH is usually single and involves the upper limb in 15% of cases. The median appearance is at age 2 weeks. When dermis is involved, the skin appears pink. Deeper lesions may cause a pale, bluish discoloration (Fig. 31.1). It grows very fast during the first 9 months (proliferating phase), begins to shrink and fade by 12 months (involuting phase), and involution is complete by 5 years (involution phase) leaving residual telangiectasias, fibrofatty residuum and redundant, atrophied skin.25,29,30
Diagnosis/patient presentation
Some 90% of IH diagnoses are made by history and physical examination. Hand-held Doppler shows fast flow. Ultrasonography shows fast flow, increased resistance and increased venous drainage.27 On MRI scans, hemangiomas are isointense on T1, hyperintense on T2, and enhance during proliferating phase. Rarely, is biopsy needed. Immunostaining for GLUT1, a glucose transporter specific to IH, will differentiate this lesion.31
Treatment/surgical technique
Problematic lesions too large to treat with intralesional corticosteroid injections are first managed with oral prednisone started at 3 mg/kg per day for 1 month and then tapered until it is discontinued 10–12 months later.30 Complications of long-term corticosteroid therapy are well known and usually disappear with discontinuation of therapy. Recently, propranolol has been used but its efficacy and safety compared to corticosteroids has not been established.32,33 Alternatively, the child may be switched to vincristine; interferon is no longer used in those under 12 months of age, due to spastic diplegia.
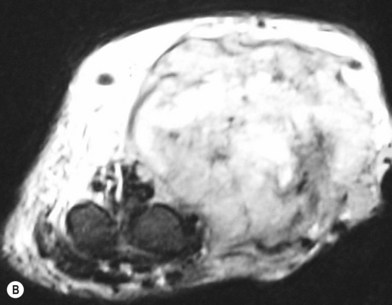
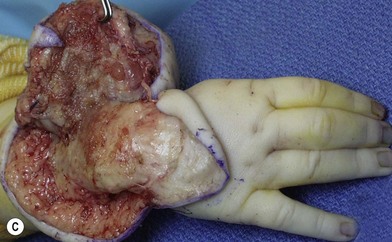
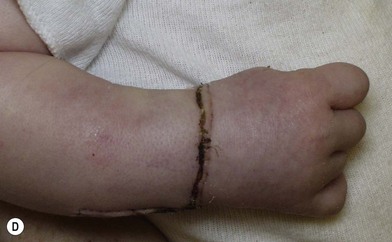
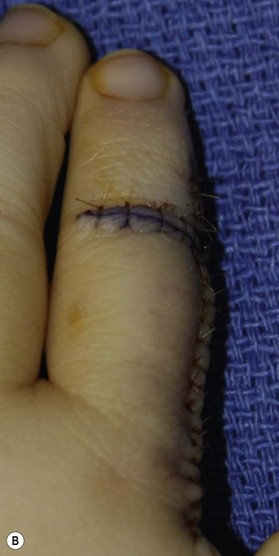
Surgery in the upper limb is indicated only for problematic ulcerations or large lesions interfering with function.2 The particular child in Figure 31.2 has a large lesion, enveloping all neurovascular structures and chronically ulcerated. Excision and neuroplasty through a high mid-axial incision both enabled functional use of the digit and avoided secondary contracture of the digit.
Congenital hemangioma (CH)
Basic science/disease process
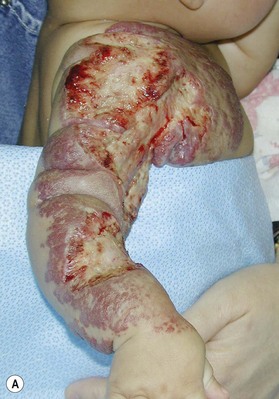
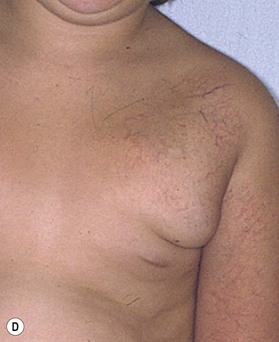
The upper limb surgeon must be familiar with a variation known as CH, which in contrast to IH are rare lesions fully grown at birth and do not exhibit the proliferation and regression so typical of IH. These lesions appear different with a red-violaceous color, course telangiectasias, a central pallor and a peripheral halo. There are two variations of CH: the rapid involuting congenital hemangioma (RICH) and the noninvoluting congenital hemangioma (NICH) (Fig. 31.3). Both lesions are commonly seen in the extremities and have an equal sex distribution. RICH involutes rapidly after birth; 50% is gone by 7 months and are completely involuted by 14 months of age.34,35,36 Fibrofatty deposits are not present. NICH does not regress and persists as raised, lumpy, plaque-like lesions with a characteristic peripheral halo. A persistent fast flow component remains unchanged.37
Pyogenic granuloma
Basic science/disease process
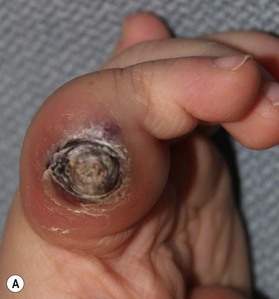
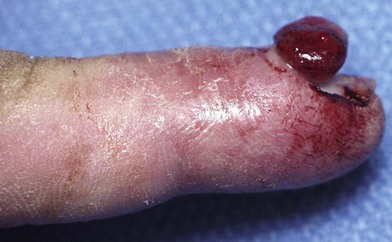
Pyogenic granuloma (PG) is a solitary red papule that grows rapidly, forming a stalk. It has been called a lobular capillary hemangioma.37 It is small with an average diameter of 6.5 mm. The male : female ratio is 2 : 1. It commonly involves skin and in the upper extremity is most problematic on the glabrous surfaces of the digits, in nail folds (Fig. 31.4), within web spaces and flexion creases. They appear as red papules growing on the end of a stalk. Persistent bleeding and constant ulceration are common. PG is distributed all over the body including the mucous membranes; cheeks, lips, oral cavity, eyelids and forehead are most common. Histological examination will differentiate PG from hemangiomas.38
Diagnosis/patient presentation
Diagnosis is made by physical examination because the appearance is classic.
Vascular malformations
Capillary malformations
Basic science/disease process
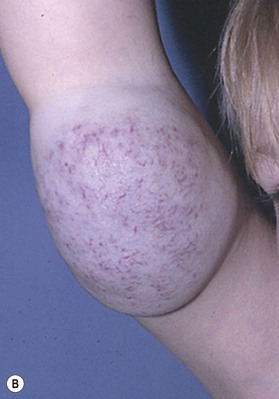
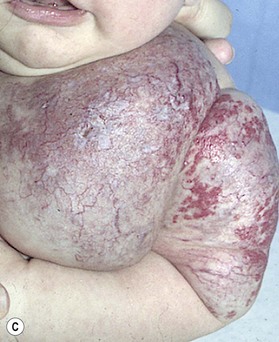
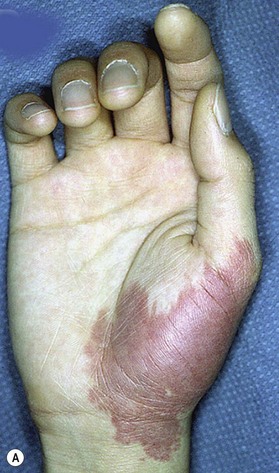
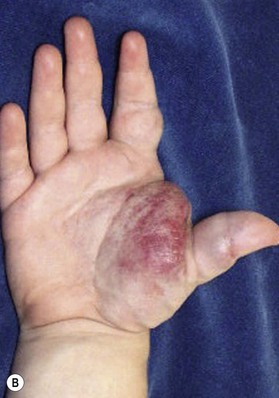
Capillary malformation (CM) consists of dilated capillaries in the superficial dermis. CM is most often solitary, but can be small or extensive and may occur in any region of the extremity (Fig. 31.5A,B) (Fig. 31.5C–F). With time and aging, these pink lesions will often darken and develop some fibrovascular overgrowth. They can be associated with deeper vascular lesions both slow-flow (Klippel–Trenaunay, CLOVES) and fast-flow (Parkes-Weber, CM-AVM, PHTS) (see Tables 31.2, 31.8) and with skeletal and soft tissue hypertrophy. A CM on the face in the one or more of the trigeminal nerve distributions is indicative of the Sturge–Weber syndrome. These patients are at risk for seizures, retinal detachment, glaucoma and stroke.
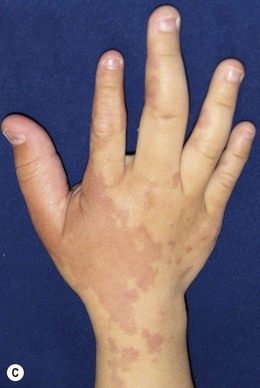
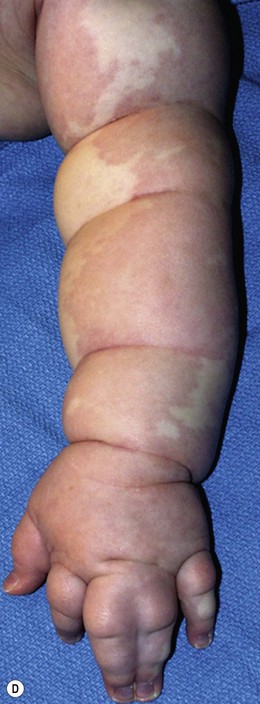
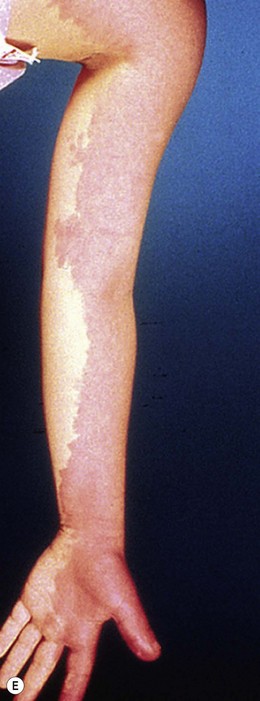
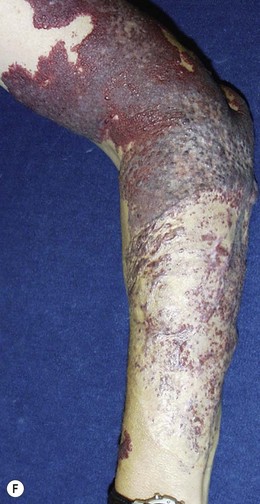
Treatment/surgical technique
No treatment is necessary for extremity CMs. Tunable pulsed-dye laser (585 mm) can effectively decrease the intensity of the cutaneous blush.39,40 Patients including infants are often treated awake and with topical anesthesia. More dramatic outcomes are obtained in the head and neck region and face than in the trunk and extremities. Multiple treatments are necessary. Outcomes in younger children are better than adults; results are variable 90% lightening of lesions, 50–90% improvement, 20%, minimal change. Many will re-darken with time.41 Surgery in these extremities is usually focused upon the deeper venous, lymphatic, combined or fast-flow lesions. Occasionally, a dark purple, overgrown extremity CM will be resected or replaced with a graft.