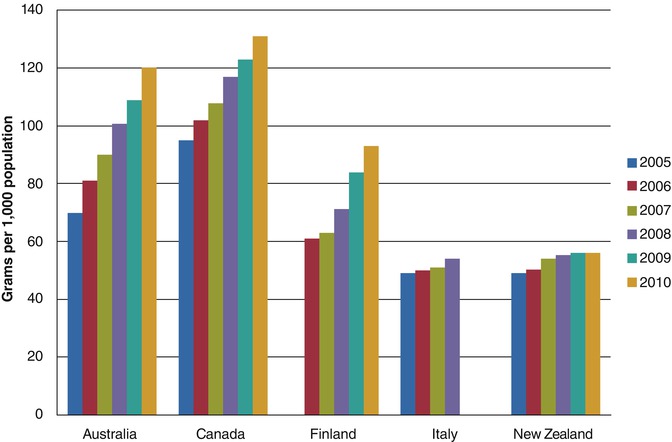
In Australia, 2003, the National Blood Agreement (NBA) was created. This led to the creation of the National Blood Authority which allows for coordination between all states and territories of Australia in policy setting, management of supply and quality and funding of blood products [3]. Demand across medical fields, including dermatology, continues to rise (Fig. 52.2) prompting the NBA to publish criteria limiting funding of IVIG to specific indications. AIBD are included in these guidelines, with funding limited to recalcitrant or relapsing cases [2, 4]. Other nations, including the USA, France and Canada, have faced similar problems with rising demands and have formed their own panels which provide guidelines on the use of IVIG. The use of IVIG in AIBD is often considered ‘off label’ by manufacturers, and funding is not routine worldwide [5–8]. For example, in the USA, the Centers for Medicare and Medicaid Services recommends funding of IVIG in patients who have failed or have contraindications to conventional therapy and states that IVIG should be used only for short-term therapy. However the definition of failing conventional therapy and what constitutes a contraindication is left to the discretion of contractors who can make access to IVIG very difficult for patients [9].
The autoimmune bullous diseases (AIBD) are a rare group of chronic, debilitating and sometimes fatal conditions affecting the skin and mucous membranes. Included in the AIBD are pemphigus, bullous pemphigoid, mucous membrane pemphigoid and epidermolysis bullosa acquisita. Mainstay of treatment is corticosteroids, the doses of which are altered according to clinical response. A significant burden of bullous diseases is due to side effects from treatment, for example, side effects from corticosteroids including immune suppression, diabetes, osteoporosis, myopathy, mood changes and peptic ulcer disease. Immunosuppressive agents, such as azathioprine and mycophenolate, are often used for their steroid-sparing effects. However these agents in turn have their own side effects including bone marrow suppression and liver function derangement. IVIG is a third-line adjunctive therapy in treatment of AIBD unresponsive to conventional therapy [10].
The action of IVIG is attributed to several mechanisms [11]:
Neutralisation of autoantibiotics
Inhibition of complement binding and activation
Fc receptor binding
Enhanced clearance of pathogenic autoantibodies via saturation of the neonatal FcR salvage pathway
Suppression of pathogenic cytokines
Downregulation of T- or B-cell function
There are many types of IVIG solution available worldwide. Differences between products include [12]:
Concentration. Usually varies between 5 and 6 % which in turn affects the volume of solution required.
Sodium and sugar (maltose or sorbitol) content. This affects osmolality and thus has effects on water retention. Low-osmolality products are more desirable for patients with conditions such as heart failure.
Plasma source. Intragam uses volunteer non-remunerated donors, Kiovig 10% uses European and USA remunerated and non-remunerated donors, and Octagam uses both remunerated and non-remunerated donors from Europe and the USA.
Plasma testing. Hepatitis B, hepatitis C and HIV are generally tested for. However, of the IVIG solutions available in Australia, only Intragam tests for human T-lymphotropic virus (HTLV). There is possibility of contraction of HTLV with other brands of IVIG.
Storage conditions. Once at room temperature, Intragam can be kept for 3 months, whereas Flebogamma can be kept for up to 1 year, and Octagam up to 2 years.
52.2 Pemphigus
Pemphigus is characterised by loss of adhesion between keratinocytes, giving rise to blister formation. This loss of adhesion (acantholysis) is due to autoantibodies directed against intercellular adhesion structures which gives rise to blisters. Disease activity corresponds to titres of autoantibodies, which can be measured by indirect immunofluorescence microscopy and ELISA against desmogleins 1 and 3. The location of blister formation varies. In pemphigus vulgaris (PV), blisters are located just above the basal skin layer, whereas in pemphigus foliaceus (PF), blisters occur within the granular layer of the epidermis. Other less common pemphigus subtypes include paraneoplastic and drug-induced pemphigus [10, 13, 14].
Several smaller studies have shown IVIG to be effective in PV and PF, even more so when administered in adjunct with a cytotoxic agent such as azathioprine or cyclophosphamide. It is thought that IVIG reduces serum levels of antibodies that mediate pemphigus, whilst cytotoxic agents reduce the rebound increase of the depleted antibody that can occur after administration of IVIG [15–17].
The first placebo-controlled study investigating the use of IVIG in PV used a single patient who suffered multiple relapses of PV despite steroids and adjunctive immunosuppressant and had numerous complications related to steroid use including diabetes, osteopenia, ruptured tendons and immunosuppression. In 2004, all adjuvant therapies save azathioprine were discontinued, and he was commenced on IVIG at a dose of 2 g/kg fortnightly for eight infusions. This led to dramatic improvement and allowed for prednisone to be reduced from 45 to 30 mg daily. He was then maintained on IVIG at 1 g/kg monthly for 16 months. Thereafter a formal randomised double-blind placebo-controlled crossover trial was conducted. There were two phases, each consisting of six consecutive months of either IVIG 1 g/kg or placebo infusion. During the trial, azathioprine and prednisone were continued, with the patient tapering the dose of prednisone by 5 mg decrements every fortnight if the disease became quiescent. The mean subjective patient disease score decreased significantly with IVIG compared to placebo (median overall scores 11.6 vs 20.6, p < 0.0001); pemphigus autoantibody titres improved significantly (1:80 vs 1:20, p = 0.007) as did desmoglein 1 and 3 antibody levels (126 vs 94 and 126 vs 79, respectively, p = 0.004). Prednisone was weaned to 33.7 mg daily on placebo versus 35.8 mg with IVIG (p < 0.0001) [16].
There has been one multicentre randomised placebo-controlled double-blind trial by Amagai et al. looking at pemphigus vulgaris unresponsive to prednisone doses over 20 mg/day. Twenty-one patients were given 2 g IVIG over 5 days (i.e. approximately 2.5 mg/kg for a 70 kg patient), 20 patients were given 1 g IVIG over 5 days, and 20 patients were given a placebo. There was a significant improvement clinically and in terms of decrease in circulating anti-desmoglein 3 IgG autoantibodies when comparing the 2 g and placebo groups, but not when comparing the 1 g and placebo groups. In contrast to many studies which suggest multiple treatment cycles of IVIG, Amagai et al. show that a single 5-day cycle has therapeutic benefits [18].
52.3 Bullous Pemphigoid
Bullous pemphigoid (BP) is characterised by deposits of IgG or C3 at the basement membrane zone of the skin, resulting in pruritus and subepidermal blister formation. IVIG in BP has been shown to achieve clinical and serological improvement [19, 20]. Like with pemphigus, IVIG appears to be more efficacious when given in adjunct with an immunosuppressive agent [21]. Recommended doses are 2 g/kg per monthly treatment cycle. Lower doses and longer intervals between treatments are associated with rapid recurrence of bullae. Once clinical control is achieved, it is best to gradually withdraw IVIG rather than stopping it abruptly (usually by increasing the interval between cycles whilst keeping the dosage of IVIG at each cycle constant) [4, 19]. IVIG is also useful in childhood BP. Based on two case reports in 3-month-old children, doses of IVIG at 300–400 mg/kg/day for a single course of 4–5 days are efficacious [22, 23].
52.4 Mucous Membrane Pemphigoid
Mucous membrane pemphigoid, also known as cicatricial pemphigoid, is a heterogenous group of chronic inflammatory subepithelial blistering diseases with dominant involvement of the mucous membranes (including ophthalmic, oral, laryngeal and genital membranes). There is linear deposition of IgG, IgA or C3 along the epithelial basement membrane zone [24].
One non-randomised study of 16 MMP patients demonstrated a significant advantage of giving IVIG compared to conventional immunosuppressants with or without steroids. IVIG at 2 g/kg per 2–4-week cycle allowed for cessation of conventional therapies without relapse in disease. There was also much more rapid control of ocular inflammation with IVIG compared to conventional treatment, suggesting that ocular MMP (which can lead to blindness) should be an indication for IVIG treatment [25]. IVIG has also been shown to be efficacious in several other case studies and trials at doses of 1–3 g/kg per cycle (generally monthly cycles) in patients pretreated with corticosteroids and/or immunosuppressants [26–28]. In one successful case study, the IVIG was given in conjunction with dapsone as the patient wished to conceive—whilst on IVIG, the patient delivered a healthy child [29]. Sometimes IVIG does not help in halting disease progression even when combined with conventional therapies [30], and treatment sometimes must be discontinued due to side effects [31].
Combination of rituximab at 375 mg/m2 (initially administered weekly for 8 weeks, then monthly for 4 months) and IVIG at 2 g/kg per monthly cycle has been successful in halting progression of blindness in patients resistant to other therapies. The IVIG is postulated to perform two functions in such a setting—firstly, helping recover B-cell levels which drop with rituximab treatment and, secondly, decreasing production of pathogenic autoantibodies [32].
52.5 Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita appears predominantly on trauma-prone areas of the skin, with blister formation, scarring and milia formation. It is characterised by autoantibodies to type VII collagen [24]. There have been several case reports on the use of IVIG in EBA.
When combined with prednisone 0.5 mg/kg/day and cyclosporine 10 mg/kg/day, IVIG at 400 mg/kg/day for 4 days, repeated every 2 weeks, has resulted in clinical improvement. With continuance of the IVIG, prednisone and cyclosporine doses were able to be decreased and autoantibody titres became undetectable [33].
Lower doses of IVIG at 40 mg/kg/day for 5 days repeated ever 3–4 weeks have also been used with success when combined with prednisone, allowing for gradual weaning off from prednisone whilst remaining disease-free on IVIG as monotherapy every 6 weeks [34].
However, when IVIG is initiated as monotherapy, even at doses of 400 mg/kg/day, there are mixed results [35, 36]. If IVIG as monotherapy is successful, maintenance with subcutaneous immunoglobulin therapy is a possibility. 0.9 g/kg/month given in divided doses on 5 days of the week has been used successfully as maintenance with no local problems despite the skin fragility seen in EBA [37].
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Azathioprine
Azathioprine
 COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


