Fig. 11.1
Anchoring fibril assembly and the consequences of the major types of mutations on type VII collagen protein synthesis in DEB. The left side shows the physiology of type VII collagen and the right side shows the pathology. Stage I: proα[alpha]1 (VII) polypeptides are synthesized in ribosomal complex. Stage II: three of these chains assemble into a triple-helical type VII collagen molecule—homotrimers. At stages III and IV, two homotrimers form antiparallel tail-to-tail dimers with a central carboxyl-terminal overlap, and with the amino-termini outward, a portion of the NC-2 domain is removed, and the association of the monomers is stabilized by intermolecular disulfide bonds. Stages V and VI: a large number of dimer molecules assemble into anchoring fibrils, and the complete NC-1 domain keeps the adhesive property at both ends. Mature anchoring fibrils are stabilized by transglutaminase cross-links in vivo. Stage VII: premature termination codon mutations (PTC) decrease the amount of the mutated transcripts and result in truncated, nonfunctional polypeptides, which are unable to assemble into anchoring fibrils, then causing RDEB-HS. Stage VIII: missense mutations alter homotrimers formation and/or subsequent stabilization of the dimmer molecules by disulfide bonds, resulting in decreased stability and/or altered function of VII collagen, known as milder mitis type of RDEB (RDEB-M). Stage IX: glycine substitutions often happen in triple-helix region of COL7A1, affecting the correct folding and the secretion of type VII collagen, resulting in DDEB (Modified from Jarvikallio et al. [18])
11.2 Classification
The clinical subtypes and features are reviewed in detail in the chapter on dystrophic EB (see Chap. 42). Inheritance patterns may be autosomal dominant (DDEB) or autosomal recessive (RDEB) or a mixture of both. DDEB previously included two classical phenotypes: the Cockayne-Touraine type and the Pasini type [10–12]. The Cockayne-Touraine type was with hypertrophic lesions, and Pasini type with white papular lesions [12]; however, the international revised classification system for EB eliminated these terms because the clinical manifestations were not consistent with the mutations and they use generalized and localized subtypes, such as pretibial, acral, and nail only [10, 11]. RDEB has been classified into the generalized severe (the term Hallopeau-Siemens type is no longer used) (RDEB-HS), intermediate and localized [10] (Fig. 11.2). There is also a pruriginosa variant, which is most typical in DDEB, and a form known as transient bullous dermolysis of the newborn (TBDN).
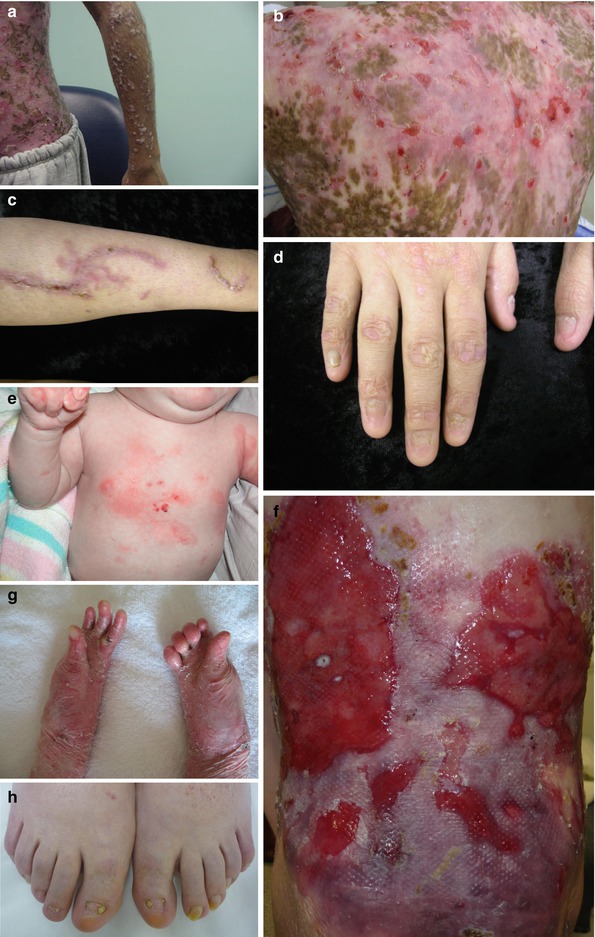
Fig. 11.2
Clinical presentation with DEB patients. (a, b) Albopapuloid lesions on the arm and 90 % of body surface covered with lesions within generalized type of DDEB (severe phenotype). (c, d) Mild lesions on the leg and severely dystrophic nails within DDEB. (e) Localized lesions to the trunk with RDEB-nGS. (f, g) Widespread blisters, erosions, scars, and atrophy and (h) significant nail dystrophy and syndactyly of the feet within GS-RDEB
11.3 Pathogenesis
RDEB-GS is generally due to premature termination codon (PTC) mutations on both COL7A1 alleles, resulting from nonsense, frameshift, or splice-site mutations [13], which result in either nonsense-mediated decay of the mRNA or truncated polypeptides that are unable to assemble into functional AF, and hence, the skin is fragile [13]. The probing of the skin with collagen VII antibodies is often negative or extremely reduced compared to normal skin. These PTC mutations are silent when in the heterozygous state, but when in the homozygous state or combined with another PTC mutation, they can result in severe RDEB (RDEB-GS). In a few cases, RDEB-GS may occur due to combinations of two missense mutations or compound heterozygosity of a missense and a PTC mutation [14].
The milder RDEB-nGS is often caused by compound heterozygous mutations, one PTC mutation and one missense mutation, and full-length type VII collagen polypeptides can be synthesized, but they have a different conformation and affect the stabilization of the AF by disulfide bonding or other structural changes [15]. Dominant dystrophic epidermolysis bullosa (DDEB) usually involves glycine substitutions within the triple helix of COL7A1 [16, 17].
11.3.1 RDEB
The milder RDEB-nGS is frequently caused by PTCs, small deletions, substitutions of glycine residues in the collagenous domain, and other missense mutations, and full-length type VII collagen polypeptides can be synthesized [18]. These mutations affect a critical amino acid and alter the conformation of the protein, which may still be able to assemble into a small number of AF but is likely to be unstable when they laterally aggregate. In these cases, probing the skin with an antibody to collagen VII reveals reduced intensity compared to normal skin. Seventeen cysteine residues are precisely conserved between the human Col7a1 and the mouse and hamster Col7a1, and one cysteine within the triple-helical domain (amino acid 2634) has been proposed to form intermolecular disulfide bonds by pairing with the first or second cysteine residue (amino acids 2802 and 2804) within the NC-2 domain of another type VII collagen molecule. Eight cysteine residues within the NC-2 domain have been suggested to participate in the formation of disulfide bonds which stabilize the antiparallel association of the two type VII collagen molecules during the extracellular assembly of AF [3, 19]. This is consistent with positive but attenuated immunohistochemical staining and reduced numbers of AF on EM in dominant and recessive DEB in which these cysteine residues are affected [11, 18]. The nature of RDEB-nGS mutations within COL7A1 is more diverse, including splice-site mutations within NC-2 [20–23], delayed termination codon (DTC) [24], in-frame exon skipping [15, 24] or missense substitution mutations involving an amino acid other than glycine [15, 25, 26], the majority involving arginine residues which result either in the loss of an ionic charge or in the introduction of a bulky chain at external positions of the triple helix [25]. TBDN may also be caused by COL7A1 mutations [27, 28] and it is not known why these usually result in transient disease. Often, on immunofluorescence microscopy, there is stippling of collagen VII in the basal cells, suggesting retention of collagen VII [29, 30].
11.3.2 Dominant Dystrophic EB (DDEB)
DDEB usually involves glycine substitutions within the triple helix of COL7A1 although other missense mutations, deletions, or splice-site mutations may underlie some cases [15–17, 31–34]. These mutations affect critical amino acids in the structure of the triple helix, and hence disrupting them may affect the overall stability of the AF. Assuming equal expression of wild-type and mutant alleles, seven-eighths of the trimeric molecules contain at least one mutant proα[alpha]1 chain, and only one-eighth consists solely of normal polypeptides [35].
More than 100 missense mutations that result in a Gly-Xaa substitution have been described in the collagenous domain of COL7A1; half of these mutations have a “dominant-negative” effect, causing DDEB, spanning from amino acids 1522–2791. A common region for mutations affects the amino acid residues 2003–2079 (exon 73–exon75) as part of a 35-triplet stretch of Gly-X-Y, which is flanked by noncollagenous sequences of 39 and 6 amino acid residues, just downstream from the 39-amino-acid hinge region (Fig. 11.3). The 35-triplet segments are evolutionarily highly conserved in the human, mouse, and hamster [8, 19]. Glycine substitution in this segment may lead to a greater destabilization than in long uninterrupted collagenous segments or close to the N- or C-terminal ends [35].
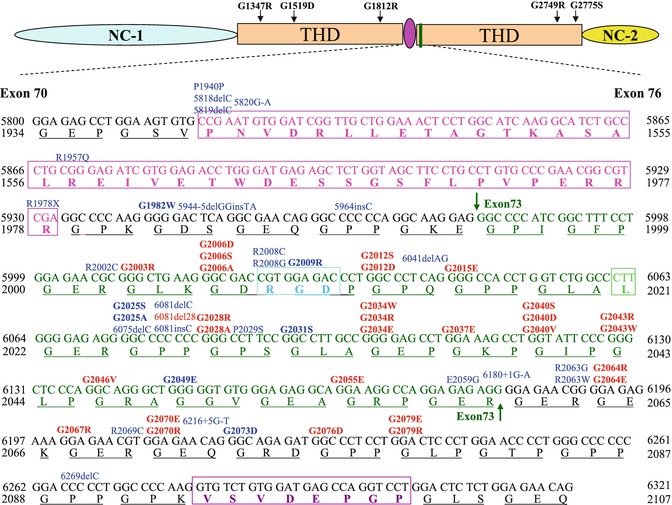
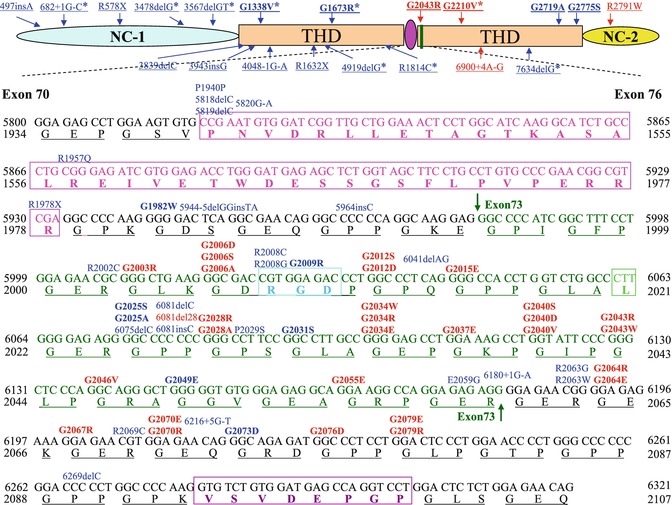
Fig. 11.3
A schematic presentation of exons 70–73 of COL7A1. The 39-amino-acid hinge region (pink) of the triple-helical domain (THD) is encoded by amino acids 1940–1978. Exon 73 is encoded by amino acid residues 1994–2060 (green), and RGD tripeptide sequence in exon 73 is outlined by a bright blue rectangle. The mutations are indicated on the top of sequences (in bold are glycine substitutions mutations); 20 dominant and 15 recessive mutations (indicated in red and blue, respectively) are clustered in exon 73, just after the 39-amino-acid interruption region. G1812R and G1519D are located within a long uninterrupted stretch of collagen VII triple helix. G2775S and G2749R are located close to C-terminal ends of the triple helix, while G1347R is closed to N-terminal ends of the triple helix
Indeed, toenail dystrophy may be the only clinical manifestation of DDEB in some families. Recent reports have shown that some glycine substitutions could lead only to nail dystrophy but no skin blistering when combined with a normal allele, such as G1815R, G1595R and G2251E, and G2287R [36, 37]. As explained below, many glycine-to-arginine substitutions in COL7A1 are “silent” according to the literature, but not all may have had their toenails well scrutinized. G2689R mutations led to an interesting phenotype, mucosal involvement, nail dystrophy, and no skin blisters [22].
In the variant of DEB known as TBDN in which there is stippling of collagen VII on IF antigen mapping without linear staining with antibodies to collagen VII [38], only four distinct mutations have been found in COL7A1, all within the collagenous domain. The first mutation was the splice mutation 4120–1G>C (IVS35–1G>C, at intron 35) [33]. The second and third mutations are two glycine substitution mutations, a novel recessive G1519D (in exon 44) and a dominant G2251E (in exon 86) mutation. The G1519D substitution is clinically silent when combined with a normal allele, and G2251E merely caused nail dystrophy in heterozygotes but no skin blistering [39]. The fourth mutation, G1522E, lies in exon 45 and was identified before in a sporadic case of EB pruriginosa [40]. G1522E and G2251E are the first amino acids encoded by exon 45 and exon 86, respectively; G1519D is close to G1522E and the last third amino acid encoded by exon 44, plus IVS35G>C; all of the four mutations are close to splice sites or in splice sites. However, Hammami-Hauasli did an analysis with G1522E using the Delila™ software package and did not predict any splice-site alterations, and IVS35G>C is predicted to create a “leaky” site, i.e., normal splicing and in-frame skipping of exon 36 [39]. The exact reason why the TBDN phenotype results from these mutations is not known, but it is proposed that they alter sites that are important for release of the polypeptide from the rough endoplasmic reticulum [40].
11.3.3 The “Silent” Glycine Substitutions in Recessive Inheritance Pattern
Not all glycine substitution mutations within COL7A1 gene are dominant. Glycine substitutions may be inherited recessively; these substitutions are usually “silent” in the heterozygous state, but when combined with another COL7A1 mutation (nonsense, splice-site, insertion, or deletion mutation), the clinical consequences are either mild, moderate, or severe RDEB [39]. It is not clear why some glycine substitutions are dominant while others are “silent” in the heterozygous state. It may be the position of these mutations within the collagenous domain of COL7A1 and the resulting degree of abnormal folding which influences the consequences at the phenotypic level. Most silent substitutions are located close to either the N- or C-terminal ends of the triple helix or in the middle of the long uninterrupted segments of Gly-X-Y repeats [35]. For example, G1812R in exon 63 is silent when combined with normal COL7A1 alleles; however, when combined with a PTC mutation on the other allele (3857delA), it results in RDEB-nHS [41]. G1519D did not interfere with folding and secretion of procollagen VII demonstrated by IF staining and immunoblot analysis; this mutation causes pathological consequences only in combination with another COL7A1 gene defect [35]. Gly-1519 and Gly-1812 are both located within a long uninterrupted stretch of collagen VII triple helix (Fig. 11.1). G2775S [42] and G2749R [14] are located close to C-terminal ends of the triple helix, while G1347R [43] is close to the N-terminal ends of the triple helix. More than half of the silent glycine substitutions are glycine-to-arginine substitutions acting in a recessive manner which have recently been reported in RDEB-nHS. Modeling studies indicate that glycine-to-arginine substitutions probably only lead to minor localized disruption of the triple-helix structure [44].
11.3.4 The De Novo Mutations
Patients with relatively mild DEB and no family history are frequently diagnosed as de novo or sporadic cases of dominant DEB, although a mild case of recessive DEB cannot be excluded on the basis of clinical and ultrastructural examination [45, 46]. The true de novo cases develop a dominant mutation in COL7A1; their offspring have a 50 % risk of inheriting the mutation. However, if one parent is a germline mosaic, the risk then depends on the percentage of mutated germline cells. If neither parent is a germline mosaic, then the risk of having another child with de novo DDEB is the same as in the general population [47]. Indeed, exon 73 has previously been shown to harbor a large number of glycine substitutions, including de novo mutations. De novo glycine substitutions resulting in DDEB are rare, but the following mutations have been reported: G1775D, G2067R [17], G2012S [48], G2012D [49], G2028R [50], G2040V [47], G2043R, G2043W [51], G2076D [52], G2079E [45], and G2348R [53]. The de novo mutations usually result from glycine substitutions and are seen in DDEB, but Posteraro et al. detected the 8117delC frameshift mutation in a patient affected with the less severe RDEB-nHS variant as a de novo mutation [17].
11.3.5 Recurrent Mutations
Although most COL7A1 mutations have been specific to the individual families, with no “hotspot” mutations, some recurrent mutations have been found in certain ethnic backgrounds. In Italy, six recurrent mutations were found including 497insA, 4738G-A, 7344G-A, 425A-C, G1664A, and 8441–14del21 [48]. The mutation 2470insC has only been found in Mexico [54]. R578X, 7786delG, and R2814X mutations are specifically limited to British patients, and 5818delC, 6573+1G-C, and E2857X are frequent in Japanese [55]. A high recurrence of 425A-G was found in central European patients [56].
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


