Urticaria and Angioedema: Introduction
|
Urticaria is defined as a skin lesion consisting of a wheal-and-flare reaction in which localized intracutaneous edema (wheal) is surrounded by an area of redness (erythema) that is typically pruritic. Individual hives can last as briefly as 30 minutes to as long as 36 hours. They can be as small as a millimeter or 6–8 inches in diameter (giant urticaria). They blanch with pressure as the dilated blood vessels are compressed, which also accounts for the central pallor of the wheal. The dilated blood vessels and increased permeability that characterize urticaria are present in the superficial dermis and involves the venular plexus in that location. Angioedema can be caused by the same pathogenic mechanisms as urticaria but the pathology is in the deep dermis and subcutaneous tissue and swelling is the major manifestation. The overlying skin may be erythematous or normal. There is less pruritus (fewer type C nerve endings at the deeper cutaneous levels) but there may be pain or burning.
Epidemiology
Urticaria and angioedema are common. Age, race, sex, occupation, geographic location, and season of the year may be implicated in urticaria and angioedema only insofar as they may contribute to exposure to an eliciting agent. Of a group of college students, 15%–20% reported having experienced urticaria, while 1%–3% of the patients referred to hospital dermatology clinics in the United Kingdom noted urticaria and angioedema. In the National Ambulatory Medical Care Survey data from 1990 to 1997 in the United States, women accounted for 69% of patient visits. There was a bimodal age distribution in patients aged birth to 9 years and 30–40 years.1
Urticaria/angioedema is considered to be acute if it lasts less than 6 weeks. Most acute episodes are due to adverse reactions to medications or foods and in children, to viral illnesses. Episodes of urticaria/angioedema persisting beyond 6 weeks are considered chronic and are divided into two major subgroups: (1) chronic autoimmune urticaria (45%) and (2) chronic idiopathic urticaria (55%) with a combined incidence in the general population of 0.5%.2 Physically induced urticaria/angioedema is not included in the definition. Various types of physical urticaria/angioedema may last for years, but the individual lesions last fewer than 2 hours (except delayed pressure urticaria) and are intermittent. Whereas 85% of children experience urticaria in the absence of angioedema, 40% of adult patients with urticaria also experience angioedema.
Approximately 50% of patients with chronic urticaria (with or without angioedema) are free of lesions within 1 year, 65% within 3 years, and 85% within 5 years; fewer than 5% have lesions that last for more than 10 years. Angioedema alters the natural history, and only 25% of patients experience resolution of lesions within 1 year. There are no data regarding the remission rate in patients with only angioedema. The hereditary group is considered to be life long once the diagnosis becomes clinically manifest.
Pathogenesis
The mast cell is the major effector cell in most forms of urticaria and angioedema, although other cell types undoubtedly contribute. Cutaneous mast cells adhere to fibronectin and laminin through the very late activation (VLA) β1 integrins VLA-3, VLA-4, and VLA-5 and to vitronectin through the αvβ3 integrin. Cutaneous mast cells, but not those from other sites, release histamine in response to compound 48/80, C5a, morphine, and codeine. The neuropeptides substance P (SP), vasoactive intestinal peptide (VIP), and somatostatin, (but not neurotensin, neurokinins A and B, bradykinin, or calcitonin gene-related peptide), activate mast cells for histamine secretion. Dermal microdialysis studies of the application of SP on skin indicate that it induces histamine release only at 10−6 M, which suggests that after physiologic nociceptor activation, SP does not contribute significantly to histamine release.3 Yet it is a major contributor to the flare reaction induced by histamine stimulation of afferent type C fibers (mediating pruritus) with release of SP from adjacent nerve endings by antidromic conduction. Histamine is found associated with the wheal.4 Recently, the spinal cord afferent fibers mediating pruritis have, for the first time, been distinguished from pain fibers in the lateral spinothalamic tracts.5
Not all potential biologic products are produced when cutaneous mast cells are stimulated. For example, SP releases histamine from cutaneous mast cells above 10−6 M but does not generate prostaglandin D2 (PGD2). Vascular permeability in skin is produced predominantly by H1 histamine receptors (85%); H2 histamine receptors account for the remaining 15%.
The current hypothesis regarding cellular infiltration that follows mast cell degranulation suggests that the release of mast cell products (histamine, leucotrienes, cytokines, chemokines) leads to alterations in vasopermeability, upregulation of adhesion molecules on endothelial cells, and rolling and attachment of blood leukocytes, followed by chemotaxis and transendothelial cell migration.
Various forms of physical urticaria/angioedema have provided experimental models for the study of urticaria/angioedema by allowing the observation of the elicited clinical response, examination of lesional and normal skin biopsy specimens, assay of chemical mediators released into the blood or tissues, and characterization of peripheral leukocyte responses.6,7 The intracutaneous injection of specific antigen in sensitized individuals has provided an experimental model for analysis of the role of immunoglobulin (Ig) E and its interaction with the mast cell. In many subjects, the challenged cutaneous sites demonstrate a biphasic response, with a transient, pruritic, erythematous wheal-and-flare reaction followed by a tender, deep, erythematous, poorly demarcated area of swelling that persists for up to 24 hours. This is the late-phase response with recruitment of variable numbers of neutrophils, prominent eosinophils, monocytes, small numbers of basophils, and CD4+ T-lymphocytes of the TH2 subclass.8 Chemokines (chemotactic cytokines) strongly associated with Th2 lymphocyte predominance include those reactive with chemokine receptors CCR3, CCR4, and CCR8 on T lymphocytes. Characteristic cytokines produced by Th2 lymphocytes include interleukins (ILs) 4, 5, 9, 13, 25, 31 and 33. The cellular infiltrate seen in biopsy specimens of delayed pressure urticaria is a variant of a late-phase reaction while mast cell degranulation in most other physical urticarias has no associated late phase. These include typical acquired cold urticaria, cholinergic urticaria, dermatographism, and type I solar urticaria.
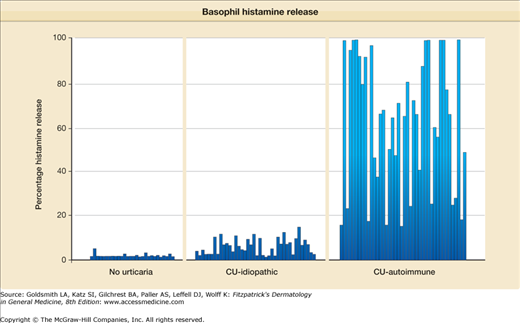
The first suggestion that patients with chronic urticaria and angioedema might have an autoimmune diathesis was the observation that there is an increased incidence of antithyroid antibodies in such patients relative to the incidence in the population at large.9 These include antimicrosomal (perioxidase) and antithyroglobulin antibodies, as seen in patients with Hashimoto’s thyroiditis.10 Patients may have clinical hypothyroidism, but a small number might be hyperthyroid if inflammation is at an early stage when thyroid hormone is released into the circulation. This atypical presentation should be distinguished from the occasional patient with Grave’s disease. Nevertheless, most patients are euthyroid. The incidence of antithyroid antibodies in chronic urticaria, as reported in the literature, varies between 15% and 24%,11,12 but the most recent data are closer to the latter figure12 and demonstrate segregation of antithyroid antibodies with chronic autoimmune urticaria rather than chronic idiopathic urticaria. However, the association is not absolute. The incidence in the autoimmune subgroup was 27%, in the chronic idiopathic urticaria subgroup 11%, while in the population at large it is 7%–8%. Gruber et al (1988)13 considered the possibility that patients might have circulating and anti-IgE antibodies that are functional and did indeed find these in about 5%–10% of patients. Gratten et al14,15 sought antibodies reactive with skin mast cells by performing an autologous skin test and found a 30% incidence of positive reactions in patients with chronic urticaria. There were only rare positive reactions in healthy control subjects or patients with other forms of urticaria. Subsequently, this level of positivity was shown by Hide et al16 to be due to an IgG antibody reactive with the α subunit of the IgE receptor; in addition a 5%–10% incidence of functional anti-IgE antibodies was confirmed (eFig. 38-1.1).17
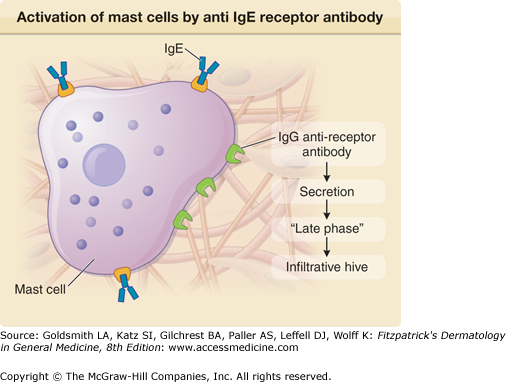
Assays for anti-IgE receptor antibody have been included in vivo methods and in vitro assays. The autologous skin test,15,18 as noted above, provided one of the first clues that a subset of patients with chronic urticaria have an autoimmune disorder. Basophils are frequently used as an in vitro surrogate for mast cells, and secretion of histamine as a result of incubation with patient sera or purified IgG was readily demonstrated,19–21 but was not observed if the source of basophils was from a “nonreleaser,” i.e., basophils with an abnormality in the signal transduction molecules lyn or syk, which are unresponsive to signals through the IgE receptor but are normally responsive to other secreteagogues, such as the cytokine monocyte chemoattractant protein-1.22 The incidence of a positive assay result was generally higher than that observed with the autologous skin test; reported values varied between 35% and 50%.19,20,23 Absorption of sera, with cloned α subunit, decreased the percentage of histamine release as the amount of added α subunit was increased22 confirming reactivity with this receptor subunit. The assay could be made more sensitive by preincubating basophils with IL-3,24 although the percentage of positive reactions was not significantly affected. Release of leucotriene C4 and IL-3 along with histamine was also demonstrable.25,26 A representative assay is shown in Fig. 38-1. Although more cumbersome, activation of cutaneous mast cells could also be performed in vitro by using either thin skin slices27 or partially purified mast cells derived from foreskin samples.28
Attempts to quantitate IgG antibody to the α subunit have, in general, been unsuccessful. Initial attempts to demonstrate the presence of antibody by means of immunoblotting23,29 have not proved useful because positive reactions can be seen in other autoimmune disorders29 or occasionally with sera23 from subjects with no history of urticaria. In addition, studies of the relationship of a positive immunoblot result with histamine release did not yield a significant correlation;22 sera with a positive blot result but negative histamine release were frequently seen. However, sera with positive histamine release and a negative blot result did have demonstrable IgG anti-α because absorption of such sera with the α subunit inhibited histamine release. Thus, lack of sensitivity of the immunoblot explained part of the discrepancy, but the cause of the false-positive blot results was not apparent. The latest observations germane to this enigma relate to the fact that the α subunit employed is cloned in an insect vector, thus glycosylation differs from that in humans; also traces of insect protein may be present. Antibody to either of these would interfere with immunoblots (glycosylation) or ELISA assays (glycosylation or contaminant).30
Subclass analysis of the pathogenic IgG has been helpful in further delineating the relationship (or lack thereof) of binding assays versus functional methods. An early report suggested that much of the antireceptor antibody exists within the IgG1 and IgG3 subclasses, but these data were based on immunoblot analysis.29 Thus, we employed affinity chromatography to purify IgG subclasses from the sera of patients with chronic urticaria to determine which IgG subclasses have functional antireceptor antibody. When we isolated IgG2 antibody from nine patients with chronic urticaria, none of them released histamine from human basophils, irrespective of whether a positive IgG2 anti-α antibody immunoblot result was seen or not. Much of the histamine release does appear to be due to IgG1 and IgG3, and rarely due to IgG4.31
Recently, a series of publications have demonstrated that normal serum has natural antibody to the α subunit of the IgE receptor with germ line variable region sequences that is primarily IgM.32–34 Such antibodies can be functional but appear to require stripping of IgE from donor basophils. Evidence to suggest cross-reactivity of anti-α antibody with tetanus toxoid was reported, which implied immunization as a cause of such antibodies; however, we have been unable to absorb sera with tetanus toxoid and diminish histamine release (A. Kaplan, unpublished observations), and if such antibodies are part of our innate repertoire, no external stimulus is needed to explain their presence. But some unknown stimulus or abnormal control mechanism might lead to pathogenic IgG antibody, such as we have seen.
That complement might contribute to the histamine release observed with sera from patients with chronic urticaria was suggested by studies in which complement depletion or inactivation appeared to diminish histamine release.21,29 A series of reports by Ferrer et al28 and Kikuchi and Kaplan22,35 then documented not only a role for complement but also more specifically activation of the classical pathway and generation of C5a. First, we demonstrated that the addition of purified patient IgG to normal serum (as a source of complement) but not sera deficient in C2 or C5 augmented cutaneous mast cell histamine release.28 This was confirmed with basophils.22,35 We could also reconstitute C5-deficient serum with purified C5 to recover the augmentation of histamine release provided by serum. Then we demonstrated inhibition of the complement contribution to histamine release by using an antibody to the C5a receptor.35 It is not clear why the presence of a functional anti-IgE receptor would cause symptoms limited to the skin, but among the differences between pulmonary and cutaneous mast cells is the absence of C5a receptors on lung mast cells. On the other hand, it is also not clear why the IgG2 anti-IgE receptor antibody, which is often demonstrable by means of immunoblotting, does not activate basophils or cutaneous mast cells because IgG2 anti-FcϵRIα should cross-link α subunits, as might any IgG subclass antibody directed to the α subunit. The difference might have to do with antibody affinity or differing α subunit epitopes (including the aforementioned nonhuman carbohydrate linkage) with which these antibodies react. The geometry of binding is also a factor because the Fc region of two adjacent IgG molecules must each be able to bind to one of the globular heads of C1Q to initiate complement activation (Fig. 38-2). It is theoretically possible to have binding of IgG to the IgE receptor in the absence of cross-linking (e.g., the receptors are too far apart) and still observe histamine release if the two Fc regions are sufficiently close together to activate C1. If receptors are sparse, the antibody can bind without activating either the cells or complement, whereas receptors saturated with IgE might preclude binding. These considerations may account for the marked heterogeneity in patient manifestations and severity.
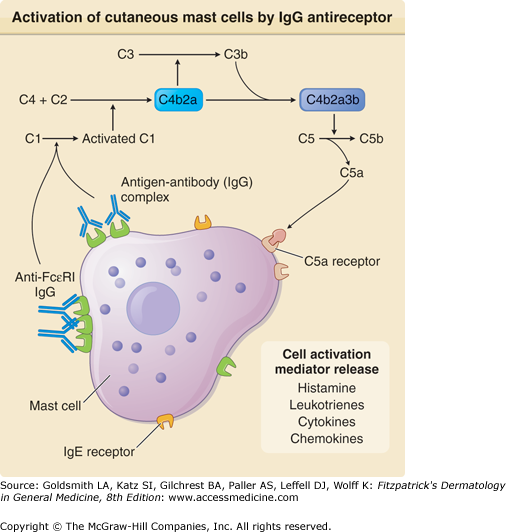
Mast cell degranulation certainly initiates the inflammatory process in autoimmune chronic urticaria and is assumed to also do so in idiopathic chronic urticaria. Evidence for an increased number of mast cells in chronic urticaria has been presented,36,37 but there are also publications indicating no significant differences from normal;38 these studies did not discriminate the autoimmune from the idiopathic groups. However, no alternative mechanisms for mast cell degranulation in the idiopathic groups have been suggested to date. Yet the histology of the two groups differs only in minor ways. Common to all biopsy specimens is a perivascular infiltrate that surrounds small venules within the superficial and deep venular plexus, with a prominence of CD4+ T lymphocytes and monocytes and virtually no B cells.36,39 Granulocytes are quite variable but are plentiful if the lesion undergoes biopsy early in its development. Neutrophils and eosinophils are both present,40,41 although the degree of eosinophils accumulation varies greatly.39 Even when eosinophils are not evident, major basic protein can be identified within lesions (in at least two-thirds of patients), which most likely represents evidence of prior eosinophil degranulation.42 The presence of basophils has also been recently demonstrated by using an antibody (BB1) that is specific for this cell type.41 Thus, the infiltrate resembles that of an allergic late-phase reaction, as suggested previously,43 although the percentage of each cell types differs, with neutrophils and monocytes being relatively more prominent in urticaria. Endothelial cell activation is suggested by the presence of intercellular adhesion molecule 1 and E-selectin in biopsy specimens of urticarial lesions.44 Sources of chemokines include the mast cell and the activated endothelial cell; the latter cells are stimulated not only by cytokines or monokines, such as IL-4, IL-1, and tumor necrosis factor-α (TNF-α), but also by the vasoactive factors, for example, histamine and leukotrienes released from activated mast cells.45 Complement activation and the release of C5a results not only in augmented mast cell (and basophil) histamine release, but C5a is also chemotactic for neutrophils, eosinophils, and monocytes. The presence of C5a is one of the factors that would distinguish this lesion from a typical allergen-induced cutaneous late-phase reaction. The particular chemokines released in chronic urticaria have not been studied. The presence of increased plasma IL-4 levels25 in patients with chronic urticaria provides indirect evidence of lymphocyte activation, basophil activation, or both, and isolated CD4+ lymphocytes of patients were shown to secrete greater amounts of both IL-4 and IFN-γ compared with that seen in healthy control subjects on stimulation with phorbol myristate acetate.
A direct comparison between cutaneous late-phase reactions and the histology of chronic urticaria revealed that infiltrating cells had characteristics of both TH1 and TH2 cells, with production of IFN-γ by the former cells and IL-4 and IL-5 by the latter.46 Alternatively, this might represent activated TH0 cells (i.e., activated CD4+ lymphocytes that are not differentiated to TH1 or TH2 cells). When the histology of autoimmuneand idiopathic chronic urticarias was compared,41 the autoimmune subgroup had greater prominence of granulocytes within the infiltrate, whereas other infiltrating cells were quite similar, with a small increment in cytokine levels in the autoimmune group and greater tryptase positivity (? less degranulation) in the autoantibody-negative group. The patients with autoimmune chronic urticaria generally had more severe symptoms than those with idiopathic chronic urticaria.47
(Figs. 38-1 and 38-2)
The basophils of patients with chronic urticaria have been shown to be hyporesponsive to anti-IgE, an observation made by Kern and Lichtenstein48 long before there were any clues to the pathogenesis of this disorder. These findings were confirmed49 and appeared to be associated with basopenia50 and to segregate with the autoimmune subgroup. One obvious interpretation is that there is in vivo desensitization of basophils in the presence of circulating anti-IgE receptor. Vonakis et al have demonstrated that patients’ basophil hyporesponsiveness to anti-IgE is due to augmented levels of SHIP phosphatase51 that limits phosphorylation reactions critical for histamine secretion. Although manifest in about half the patients with chronic urticaria (and not segregated with either the autoimmune or idiopathic subgroups), the abnormality appears to reverse when patients remit. Thus, it may be a marker of disease activity. We have found a paradoxical result when the isolated basophils of patients with chronic urticaria were activated and compared with the basophils of healthy control subjects. Although the basophils of the patients with urticaria were clearly less responsive to anti-IgE, they demonstrated augmented histamine release when incubated with serum and it did not matter whether the sera were taken from normal subjects, other patients with chronic urticaria, or was their own.52
Studies of the plasma of patient with chronic urticaria demonstrate the presence of d-dimer and prothrombin 1 and 2 fragments indicating activation of prothrombin to thrombin as well as digestion of fibrinogen by thrombin.53 The reaction is not specific for chronic urticaria as similar observations have been noted in multiple nonsteroidal hypersensitivity syndrome.54 Nevertheless, the data are of considerable interest and activation of the coagulation cascade is dependent on tissue factor rather than factor XII, i.e., the extrinsic coagulation cascade. Although activated endothelial cells are a well-known source of the tissue factor, histologic studies suggest that eosinophils are a prominent source.55 The relationship of these observations to histamine release by basophils or mast cells is not clear. Whereas thrombin activation of mast cells has been reported, the amounts required are large and the observations thus far are confined to rodent mast cells. One publication relating to eosinophil to histamine release found IgG antibody to FceRII in the serum of patients with chronic urticaria which activates eosinophils to release cationic proteins.56 They propose basophil activation by these eosinophil cationic proteins but do not demonstrate it; however, they offer an additional mechanism for basophil and possibly mast cell histamine release.
Kinins are low-molecular-weight peptides that participate in inflammatory processes by virtue of their ability to activate endothelial cells and, as a consequence, lead to vasodilatation, increased vascular permeability, production of nitric oxide, and mobilization of arachidonic acid. Kinins also stimulate sensory nerve endings to cause a burning dysesthesia. Thus, the classical parameters of inflammation (i.e., redness, heat, swelling, and pain) can all result from kinin formation. Bradykinin is the best characterized of this group of vasoactive substances.
There are two general pathways by which bradykinin is generated. The simpler of the two has only two components: (1) an enzyme tissue kallikrein57 and (2) a plasma substrate, low-molecular-weight kininogen.58,59 Tissue kallikrein is secreted by many cells throughout the body; however, certain tissues produce particularly large quantities. These include glandular tissues (salivary and sweat glands and pancreatic exocrine gland) and the lung, kidney, intestine, and brain.
The second pathway for bradykinin formation is far more complex and is part of the initiating mechanism by which the intrinsic coagulation pathway is activated (eFig. 38-1.2).60 Factor XII is the initiating protein that binds to certain negatively charged macromolecular surfaces and autoactivates (autodigests) to form factor XIIa.61,62 This is synonymous with Hageman factor as designated in the figure. There are two plasma substrates of factor XIIa, namely (1) prekallikrein63 and (2) factor XI,64,65 and each of these circulates as a complex with high-molecular-weight kininogen (HK).66,67 These complexes also attach to initiating surfaces, and the major attachment sites are on two of the domains of HK, which thereby places both prekallikrein and factor XI in optimal conformation for cleavage to kallikrein (plasma kallikrein) and factor XIa, respectively. It is important to note that plasma kallikrein and tissue kallikrein are separate gene products and have little amino acid sequence homology, although they have related functions (i.e., cleavage of kininogens). Tissue kallikrein prefers low-molecular-weight kininogen but is capable of cleaving HK, whereas plasma kallikrein cleaves HK exclusively. The two kininogens have an identical amino acid sequence starting at the N-terminus and continuing to 12 amino acids beyond the bradykinin moiety59 but differ in C-terminal domains because of alternative splicing at the transcription level.68,69 Both factor XII and HK bind to endothelial cells (which may function as the “natural” surface in the presence of physiologic zinc ion), thus activation may occur at the cell surface.70,71
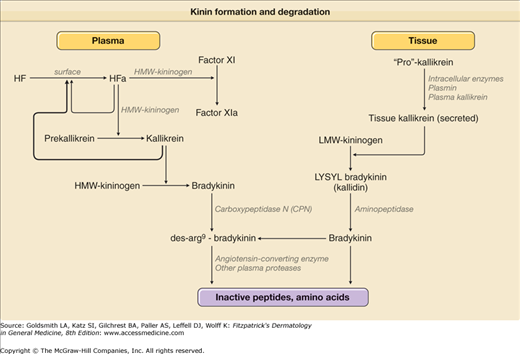
A scheme for both production and degradation of kinins is shown in eFig. 38-1.2. The enzymes that destroy bradykinin consist of kininases I and II. Kininase I is also known as plasma carboxypeptidase N,72 which removes the C-terminal arg from bradykinin or kallidin to yield des-arg73 bradykinin or des-arg74 kallidin, respectively.75 It is the same enzyme that cleaves the C-terminal arg from the complement anaphylatoxins C3a and C5a. Kininase II is identical to angiotensin-converting enzyme (ACE).76 Kininase II is a dipeptidase that cleaves the C-terminal phe-arg from bradykinin to yield a heptapeptide, which is cleaved once again to remove ser-pro and to leave the pentapeptide arg-pro-pro-gly-phe.75 If the C-terminal arg of bradykinin is first removed with kininase I, then ACE functions as a tripeptidase to remove ser-pro-phe and to leave the above pentapeptide.77 Bradykinin and kallidin stimulate constitutively produced B2 receptors,78 whereas des-arg73-BK or des-arg74 lys-BK both stimulate B1 receptors,79 which are induced as a result of inflammation. Stimuli for B1 receptor transcription include IL-1 and TNF-α.80,81
Clinical Findings
Circumscribed, raised, erythematous, usually pruritic, evanescent areas of edema that involve the superficial portion of the dermis are known as urticaria (Fig. 38-3); when the edematous process extends into the deep dermis and/or subcutaneous and submucosal layers, it is known as angioedema. Urticaria and angioedema may occur in any location together or individually. Angioedema commonly affects the face or a portion of an extremity, may be painful but not pruritic, and may last several days. Involvement of the lips, cheeks, and periorbital areas is common, but angioedema also may affect the tongue, pharynx, or larynx. The individual lesions of urticaria arise suddenly, rarely persist longer than 24–36 hours, and may continue to recur for indefinite periods. They are highly pruritic.
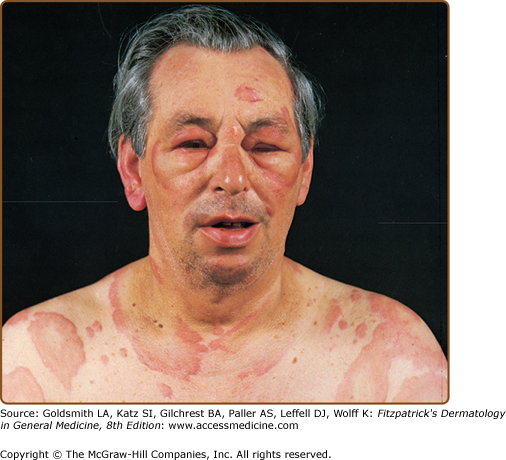
Episodes of acute urticaria/angioedema that occur in individuals with a personal or family history of asthma, rhinitis, or eczema are presumed to be IgE dependent. However, in clinical practice, urticaria/angioedema infrequently accompanies an exacerbation of asthma, rhinitis, or eczema. The prevalence of chronic urticaria/angioedema is not increased in atopic individuals.
Common examples of specific antigens that provoke urticaria/angioedema include foods such as shellfish, nuts, and chocolate; drugs and therapeutic agents notably penicillin; aeroallergens; and Hymenoptera venom (see Fig. 38-3). Urticaria in patients with helminthic infestations has been attributed to IgE-dependent processes; however, proof of this relationship is often lacking. Specific allergens and nonspecific stimuli may activate local reactions termed recall urticaria at sites previously injected with allergen immunotherapy.
Dermographism is the most common form of physical urticaria and is the one most likely to be confused with chronic urticaria. A lesion appears as a linear wheal with a flare at a site in which the skin is briskly stroked with a firm object (Fig. 38-4). A transient wheal appears rapidly and usually fades within 30 minutes; however, the patient’s normal skin is typically pruritic so that an itch–scratch sequence may appear. The prevalence of dermographism in the general population was reported as 1.5% and 4.2%, respectively, in two studies, and its prevalence in patients with chronic urticaria is 22%. It is not associated with atopy. The peak prevalence occurs in the second and third decades. In one study, the duration of dermographism was greater than 5 years in 22% of individuals and greater than 10 years in 10%.
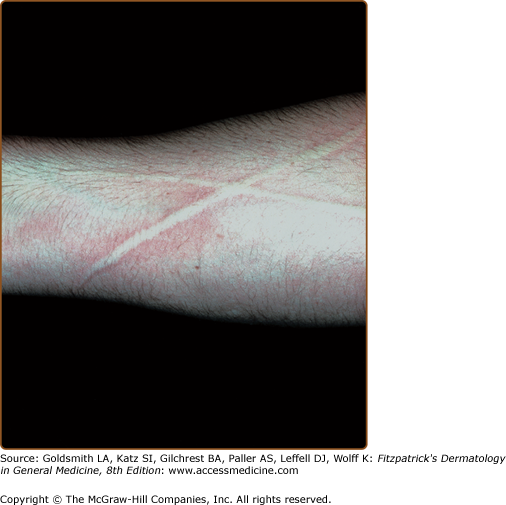
Elevations in blood histamine levels have been documented in some patients after experimental scratching, and increased levels of histamine,82 tryptase, SP, and VIP, but not calcitonin gene-related peptide, have been detected in experimental suction-blister aspirates. The dermographic response has been passively transferred to the skin of normal subjects with serum or IgE.83
In delayed dermographism, lesions develop 3–6 hours after stimulation, either with or without an immediate reaction, and last 24–48 hours. The eruption is composed of linear red indurated wheals. This condition may be associated with delayed pressure urticaria and these two may, in fact, represent the same entity. Cold-dependent dermographism is a condition characterized by marked augmentation of the dermatographic response when the skin is chilled.84
Delayed pressure urticaria appears as erythematous, deep, local swellings, often painful, that arise from 3 to 6 hours after sustained pressure has been applied to the skin.85,86 Spontaneous episodes are elicited on areas of contact after sitting on a hard chair, under shoulder straps and belts, on the feet after running, and on the hands after manual labor. The peak prevalence occurs in the third decade. Delayed pressure urticaria may occasionally be associated with fever, chills, arthralgias, and myalgias, as well as with an elevated erythrocyte sedimentation rate and leukocytosis. In one study, it accompanied chronic urticaria in 37% of patients. This is far more commonly seen than patients with pressure urticaria and no spontaneously occurring hives. An IgE-mediated mechanism has not been demonstrated; however, histamine and IL-6 have been detected in lesional experimental suction-blister aspirates and in fluid from skin chambers, respectively.87–89
Vibratory angioedema may occur as an acquired idiopathic disorder, in association with cholinergic urticaria, or after several years of occupational exposure to vibration.90 It has been described in families with an autosomal dominant pattern of inheritance.91 The heritable form often is accompanied by facial flushing. An increase in the level of plasma histamine was detected during an experimental attack in patients with the hereditary form and in patients with acquired disease.91,92 A typical symptom is hives across the back when toweling off after a shower (in the absence of dermatographism).
There are both acquired and inherited forms of cold urticaria/angioedema; however, the familial form is rare. Idiopathic or primary acquired cold urticaria may be associated with headache, hypotension, syncope, wheezing, shortness of breath, palpitations, nausea, vomiting, and diarrhea. Attacks occur within minutes after exposures that include changes in ambient temperature and direct contact with cold objects. The elicitation of a wheal after the application of ice has been called a diagnostic cold contact test (Fig. 38-5). This can be performed with thermoelectric elements with graded temperatures so that the temperature threshold for producing a wheal can be determined and a dose-response (sensitivity) in terms of stimulus duration can be readily obtained.92 If the entire body is cooled (as in swimming), hypotension and syncope, which are potentially lethal events (by drowning), may occur. In rare instances, acquired cold urticaria has been associated with circulating cryoglobulins, cryofibrinogens, cold agglutinins, and cold hemolysins, especially in children with infectious mononucleosis.93–95
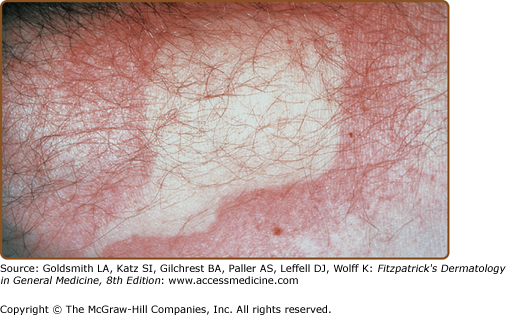
Passive transfer of cold urticaria by intracutaneous injection of serum or IgE to the skin of normal recipients has been documented.96,97 Histamine, chemotactic factors for eosinophils and neutrophils, PGD2, cysteinyl leukotrienes, platelet-activating factor, and TNF-α have been released into the circulation after experimental challenge.98–104 Histamine, SP, and VIP, but not calcitonin gene-related peptide, have been detected in experimental suction-blister aspirates. Histamine has been released in vitro from chilled skin biopsy specimens that have been rewarmed.105 Neutrophils harvested from the blood of an experimentally cold-challenged arm manifested an impaired chemotactic response suggesting in vivo desensitization. Whereas complement has no role in primary acquired cold urticaria, cold challenge of patients with cold urticaria who have circulating immune complexes (such as cryoglobulins) can provoke a cutaneous necrotizing venulitis with complement activation.106–109
Rare forms of acquired cold urticaria have been described mainly in case reports include systemic cold urticaria,84 localized cold urticaria,110 cold-induced cholinergic urticaria, cold-dependent dermographism,84 and localized cold reflex urticaria.111,112 Three forms of dominantly inherited cold urticaria have been described. Familial cold urticaria which has been termed familial cold autoinflammatory syndrome and is considered a type of periodic fever.113 It is a disorder showing an autosomal dominant pattern of inheritance with a genetic linkage to chromosomes 1q44. The responsible gene has been identified as CIASI, which codes for a protein involved in regulation of inflammation and apoptosis.114 The eruption occurs as erythematous macules and infrequent wheals and is associated with burning or pruritus. Fever, headaches, conjunctivitis, arthralgias, and a neutrophilic leukocytosis are features of attacks. The delay between cold exposure and onset of symptoms is 2.5 hours, and the average duration of an episode is 12 hours. Renal disease with amyloidosis occurs infrequently. Skin biopsy specimens show mast cell degranulation and an infiltrate of neutrophils. Results of the cold contact test and passive transfer with serum have been negative. Serum levels of IL-6 and granulocyte colony stimulating factor were elevated in one patient. Other studies suggest a pathogenic role for IL-1. Delayed cold urticaria occurs as erythematous, edematous, deep swellings that appear 9–18 hours after cold challenge. Lesional biopsy specimens show edema with minimal numbers of mononuclear cells; mast cells are not degranulated; and neither complement proteins nor immunoglobulins are detected. Cold immersion does not release histamine, and the condition cannot be passively transferred. Recently, a new form of familial cold urticaria with dominant inheritance has been reported with pruritus, erythema, and urticaria with cold exposure that can progress to syncope. The ice cube test is negative and it lacks the fever, and flu-like symptoms associated with familial cold autoinflammatory syndrome.115
Cholinergic urticaria develops after an increase in core body temperature, such as during a warm bath, prolonged exercise, or episodes of fever.116 The highest prevalence is observed in individuals aged 23–28 years. The eruption appears as distinctive, pruritic, small, 1- to 2-mm wheals that are surrounded by large areas of erythema (Fig. 38-6). Occasionally, the lesions may become confluent, or angioedema may develop. Systemic features include dizziness, headache, syncope, flushing, wheezing, shortness of breath, nausea, vomiting, and diarrhea. An increased prevalence of atopy has been reported. The intracutaneous injection of cholinergic agents, such as methacholine chloride, produces a wheal with satellite lesions in approximately one-third of patients.117,118 Alterations in pulmonary function have been documented during experimental exercise challenge119 or after the inhalation of acetylcholine, but most are asymptomatic.
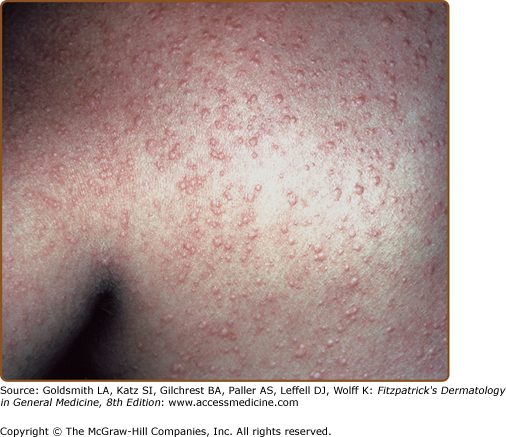
A major subpopulation of patients with cholinergic urticaria have a positive skin test result and in vitro histamine release in response to autologous sweat.120 It is not clear whether this is IgE mediated and any antigen present in sweat is unidentified. This is the same subpopulation with a positive methacholine skin test with satellite lesions and a nonfollicular distribution of the wheals. The remaining patients had negative results on autologous sweat skin tests or in vitro histamine release. Results of the methacholine skin test are negative for satellite lesions and the hives tend to be follicular in distribution.
Familial cases have been reported only in men in four families.121 This observation suggests an autosomal dominant pattern of inheritance. One of these individuals had coexisting dermographism and aquagenic urticaria.
After exercise challenge, histamine and factors chemotactic for eosinophils and neutrophils have been released into the circulation.99,119 Tryptase has been detected in lesional suction-blister aspirates. The urticarial response has been passively transferred on one occasion; however, most other attempts to do so have been unsuccessful.
Cold urticaria and cholinergic urticaria are not uncommonly seen together122,123 and cold-induced cholinergic urticaria represents an unusual variant in which typical “cholinergic” appearing lesions occur with exercise, but only if the person is chilled, for example, with exercise outside on a winter’s day. The ice cube test and methacholine skin test are both negative.124
Local heat urticaria is a rare form of urticaria in which wheals develop within minutes after exposure to locally applied heat. An increased incidence of atopy has been reported. Passive transfer has been negative. Histamine, neutrophil chemotactic activity, and PGD2 have been detected in the circulation after experimental challenge.125 A familial delayed form of local heat urticaria in which the urticaria occurred in 1–2 hours after challenge and lasted up to 10 hours has been described.
Solar urticaria occurs as pruritus, erythema, wheals, and occasionally angioedema that develop within minutes after exposure to sun or artificial light sources. Headache, syncope, dizziness, wheezing, and nausea are systemic features. Most commonly, solar urticaria appears during the third decade.126 In one study, 48% of patients had a history of atopy. Although solar urticaria may be associated with systemic lupus erythematosus and polymorphous light eruption, it is usually idiopathic. The development of skin lesions under experimental conditions in response to specific wavelengths has allowed classification into six subtypes; however, individuals may respond to more than one portion of the light spectrum. In type I, elicited by wavelengths of 285–320 nm, and in type IV, elicited by wavelengths of 400–500 nm, the responses have been passively transferred with serum, suggesting a role for IgE antibody. In type I, the wavelengths are blocked by window glass.127,128 Type VI, which is identical to erythropoietic protoporphyria, is due to ferrochelatase (hemesynthetase) deficiency (see Chapter 132).74 There is evidence that an antigen on skin may become evident once irradiated with the appropriate wave length of light followed by complement activation and release of C5a.129–131
Histamine and chemotactic factors for eosinophils and neutrophils have been identified in blood after exposure of the individuals to ultraviolet A, ultraviolet B, and visible light.132,133 In some individuals, uncharacterized serum factors with molecular weights ranging from 25 to 1,000 kDa, which elicit cutaneous wheal-and-erythema reactions after intracutaneous injection, have been implicated in the development of lesions.
Exercise-induced anaphylaxis is a clinical symptom complex consisting of pruritus, urticaria, angioedema respiratory distress, and syncope that is distinct from cholinergic urticaria.134–137 In most patients, the wheals are not punctate and resemble the hives seen in acute or chronic urticaria. The symptom complex is not readily reproduced by exercise challenges as is cholinergic urticaria. There is a high prevalence of an atopic diathesis. Some cases are food dependent, i.e., exercise will lead to an anaphylaxis-like episode only if food was ingested within 5 hours of the exercise. The food dependency is subdivided into two groups: in the first the nature of the food eaten is not relevant, whereas in the second a specific food to which there is IgE-mediated hypersensitivity must be eaten for hives to appear.138–141 Yet in these cases, eating the food without exercise does not result in urticaria. The food-dependent group is easier to treat because avoidance of food (or a specific food) for 5–6 hours before exercise prevents episodes. Cases not related to food require therapy for acute episodes and attempts to prevent episodes with high-dose antihistaminics or avoidance of exercise. Results of a questionnaire study of individuals who had had exercise-induced anaphylaxis for more than a decade142 disclosed that the frequency of attacks had decreased in 47% and had stabilized in 46%. Forty-one percent had been free of attacks for 1 year. Rare familial forms have been described. In exercise-induced anaphylaxis, baseline pulmonary function tests are normal. Biopsy specimens show mast cell degranulation, and histamine and tryptase are released into the circulation when symptoms appear.
Adrenergic urticaria occurs as wheals surrounded by a white halo that develop during emotional stress. The lesions can be elicited by the intracutaneous injection of norepinephrine.
Contact of the skin with water of any temperature may result in pruritus alone or, more rarely, urticaria. The eruption consists of small wheals that are reminiscent of cholinergic urticaria. Aquagenic urticaria has been reported in more than one member in five families.143 Aquagenic pruritus without urticaria is usually idiopathic but also occurs in elderly persons with dry skin and in patients with polycythemia vera, Hodgkin’s disease, the myelodysplastic syndrome, and the hypereosinophilic syndrome. Patients with aquagenic pruritus should be evaluated for the emergence of a hematologic disorder. After experimental challenge, blood histamine levels were elevated in subjects with aquagenic pruritus and with aquagenic urticaria. Mast cell degranulation was present in lesional tissues. Passive transfer was negative.
Urticaria may occur after direct contact with a variety of substances. It may be IgE mediated or nonimmunologic. The transient eruption appears within minutes, and when it is IgE mediated, it may be associated with systemic manifestations. Passive transfer has been documented in some instances. Proteins from latex products are a prominent cause of IgE-mediated contact urticaria.144 Latex proteins also may become airborne allergens, as demonstrated by allergen-loaded airborne glove powder used in inhalation challenge tests. These patients may manifest cross-reactivity to fruits, such as bananas, avocado, and kiwi.145 Associated manifestations include rhinitis, conjunctivitis, dyspnea, and shock. The risk group is dominated by biomedical workers and individuals with frequent contact with latex, such as children with spina bifida. Agents such as stinging nettles, arthropod hairs, and chemicals may release histamine directly from mast cells.
Papular urticari occurs as episodic, symmetrically distributed, pruritic, 3- to 10-mm urticarial papules that result from a hypersensitivity reaction to the bites of insects such as mosquitoes, fleas, and bedbugs. This condition appears mainly in children. The lesions tend to appear in groups on exposed areas such as the extensor aspects of the extremities.146
C1 inhibitor (C1 INH) is the sole plasma inhibitor of factor XIIa and factor XIIf,147,148 and it is one of the major inhibitors of kallikrein149 as well as factor XIa.150 Thus, in the absence of C1 INH, stimuli that activate the kinin-forming pathway will do so in a markedly augmented fashion; the amount of active enzyme and the duration of action of the enzymes are prolonged. C1 INH deficiency can be familial, in which there is a mutant C1 INH gene, or it can be acquired. Both the hereditary and acquired disorders have two subtypes. For the hereditary disorder, type I hereditary angioedema (HAE) (85%) is an autosomal dominant disorder with a mutant gene (often with duplication, deletions, or frame shifts) leading to markedly suppressed C1 INH protein levels as a result of abnormal secretion or intracellular degradation.151 Type 2 HAE (15%) is also a dominantly inherited disorder, typically with a point (missense) mutation leading to synthesis of a dysfunctional protein.152 The C1 INH protein level may be normal or even elevated, and a functional assay is needed to assess activity. The acquired disorder has been portrayed as having two forms, but they clearly overlap and have in common B cell activation that is often clonal. One group is associated with B-cell lymphoma153–155 or connective tissue disease,156 in which there is consumption of C1 INH. Examples are systemic lupus erythematosus and cryoglobulinemia, in which complement activation is prominent, and B-cell lymphomas, in which immune complexes are formed by anti-idiotypic antibodies to monoclonal immunoglobulin expressed by the transformed B lymphocytes.157 A second group has a prominence of a circulating IgG antibody to INH itself,158–160 but this may be seen with lymphoma or systemic lupus erythematosus as well. Acquired types have depressed C1q levels, whereas hereditary types do not, and depressed C4 levels characterize all forms of C1 INH deficiency. The acquired autoimmune subgroup has a circulating 95-kDa cleavage product of C1 INH because the antibody depresses C1 INH function yet allows cleavage by enzymes with which it usually interacts.159–162
It is now clear that depletion of C4 and C2 during episodes of swelling163,164 is a marker of complement activation but does not lead to release of a vasoactive peptide responsible for the swelling. Bradykinin is, in fact, the mediator of the swelling165–167 and the evidence in support of this conclusion is summarized below. Patients with HAE are hyperresponsive to cutaneous injection of kallikrein.168 They have elevated bradykinin levels, and low prekallikrein and HK levels during attacks of swelling.169–171 The augmentation in complement activation seen at those times may be due to activation of C1r and C1s by factor XIIf.172 The presence of kallikrein-like activity in induced blisters of patients with HAE also supports this notion,173 as does the progressive generation of bradykinin on incubation of HAE plasma in plastic (noncontact-activated) tubes165,166 as well as the presence of activated factor XII and cleaved HK levels seen during attacks.174 One unique family has been described in which there is a point mutation in the C1 INH (A1a 443 → Val) leading to an inability to inhibit complement but normal inhibition of factor XIIa and kallikrein.175,176 No family member of this type 2 mutation has had angioedema,175
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


