Ultraviolet Radiation Carcinogenesis: Introduction
|
Ultraviolet Radiation as a Carcinogen
Skin cancer offers a very clear picture of how a carcinogen causes human neoplasia. The basic principles of carcinogen exposure and slow development were discovered when Sir Percivall Pott traced scrotal cancers in adults to childhood employment as a chimney sweep1 and they also apply to sunlight-induced cancers.2,3 The process begins with carcinogen exposure, DNA damage, and failure to repair DNA or failure to apoptotically eliminate a damaged cell.4–7 A mutant gene arises in a single cell which then expands into a mutant clone.8 Rare cells of the clone repeat this carcinogenesis cycle to generate mutations in additional genes. Sunlight acts at each of these steps.
The lifetime expectation of skin cancer in Australia is ∼60%.9 In the southern United States and Hawaii, nonmelanoma skin cancers exceed all other cancers combined.10–12 Basal and squamous cell carcinomas (BCC and SCC), and an SCC precursor, actinic keratosis (AK), are most frequent on sun-exposed skin, in outdoor workers, and at lower latitudes.13–16 SCCs increase more quickly with dose of sun exposure or nearness to the equator than BCCs, and occur later in life, implying that SCC requires more sun-related steps.16–20 In contrast, one-third of BCCs occur on body sites having only intermittent sun exposure, such as the trunk and legs.16,21
Melanoma also depends on sunlight. The relation to latitude is clear,22–25 yet it is often stated that the predilection for the back and lower legs makes the relation to sunlight uncertain. This predilection likely reflects the large surface area of back and legs. When expressed as lesions per unit area, melanomas are 10–20-fold more frequent on the ears of males and face than on intermittently exposed sites such as the lower legs in women, shoulders, back, or neck.25,26 Melanomas are rare on the buttocks and soles. Melanoma appears to have two distinct origins:
A chronic sun damage (CSD) etiology affects the head and neck and is associated with chronic elastosis—a classic indicator of CSD—as well as gene amplification of the cell cycle genes CDK4 and CCND1.27 The slow-growing lentigo maligna melanoma and its precursor, lentigo maligna occur on habitually exposed body sites of light-skinned individuals.28
A non-CSD route involves intermittent sun exposure of sites such as the trunk. Non-CSD melanomas carry mutations in the BRAF or NRAS oncogene—upstream regulators of cell cycle genes—and the patients have variant alleles of the melanocortin 1 receptor.27,29,30 Intermittently exposed body sites are the main locations of melanoma increases in recent decades, melanomas in patients younger than age 50 years, and the additional melanomas seen near the equator.23,25,31 Recreational sunburn may explain these and the twofold higher melanoma incidence in office workers compared to outdoor workers.32,33
Sunlight is also implicated by the susceptible population: both classes of skin cancer are more frequent in light-skinned individuals with blonde or red hair who burn rather than tan.13,34 Compared to dark-skinned individuals, nonmelanoma skin cancer risk rises ∼10-fold in Asians and ∼100-fold in Caucasians, with a further 2–12-fold risk for people with blonde or, especially, red hair.13,34–37 The divergence is less for melanoma, about 1:1:15 (Blacks:Asians:Caucasians).38 In black skin, BCC is rare even in patients with the hereditary nevoid basal cell carcinoma syndrome (NBCCS or Gorlin syndrome).39 Skin tumors in black patients are often scar-related, but these may be associated with sunlight as well.36 Melanin-related effects result from less melanin in light skin,40 less shielding by pheomelanin than eumelanin, and greater production of photosensitized reactive oxygen from pheomelanin.41–43
Molecular epidemiology has provided the most direct evidence for ultraviolet radiation (UVR) as the active component of sunlight: UVB signature mutations are present in human BCC, SCC, AK, and melanoma (see Section “Ultraviolet Radiation-Induced Mutations”). Mutant cells are associated with elastotic dermis, indicating chronic sun exposure.44 UVB’s effectiveness is due to its ability to partially penetrate the ozone layer and stratum corneum and then be absorbed by DNA.45 The ozone layer absorbs all but 1 part per million of UVC (used in germicidal bulbs), which otherwise would be lethal; UVA penetrates well but is poorly absorbed by DNA.45,46 Nevertheless, chronic UVA can induce tumors in mice47 and malignantly transforms predisposed human cells.48 The cumulative dose of sunlight required to cause BCC or SCC in adults is fairly large, approximately 10,000 and 70,000 hours of exposure, respectively.16 Psoriasis patients who received long-term maintenance UVB phototherapy had a three- to eightfold higher risk of nonmelanoma skin cancer than people with an outdoor occupation, although the mean annual UVB dose received by psoriasis patients was 22 J/cm2, lower than the solar UVB dose annually received by individuals with an outdoor occupation (134 J/cm2).49
Some melanomas appear to be independent of sunlight: tumors of the mucosa, palms, soles, and nail beds are equally frequent in whites and blacks and have remained constant over time. In contrast, melanomas of the skin have increased manyfold in recent decades.50 Ocular melanomas are more frequent in whites than blacks,51 but have not increased in the last few decades.52,53 Although the risk of external ocular melanoma (eyelid and conjunctival melanomas) decreases with higher latitude (less sun exposure), the risk of internal ocular melanoma (uveal melanoma), which is not exposed to sunlight, increases with higher latitude, as does the risk of other internal malignancies.53,54
The incidence of melanoma and nonmelanoma skin cancers has doubled each decade since the 1960s.10,23,25,55–57 AK, lentigo maligna, and lentigo maligna melanoma—typically lesions of the middle-aged and elderly—are now seen in young adults. The best evidence that recreational sun exposure is responsible for much of the increase in skin cancers is that intermittently sun-exposed sites, such as the trunk and limbs, account for most of the increase in skin cancers, with little change in melanomas of the head and neck.10,23,25 Another suspect has been ozone depletion; because of a steep absorption curve in the UVB region, small changes in ozone concentration greatly affect UVB penetration (UVC is fortunately still blocked). Epidemiological data do not support a close relationship between ozone holes and skin cancer rates. The Antarctic ozone hole caused a 50% ozone reduction over southern Chile and Argentina in the last two decades, with UVB increasing up to 40-fold. Yet skin cancers in these areas are increasing at the same rate as elsewhere.58,59 The Arctic ozone hole has been offset by screening from air pollutants, yet skin cancers in Scandinavia are rising.60
An iatrogenic source of increased skin cancer incidence is psoralen plus ultraviolet A (PUVA) therapy for psoriasis, which increases the risk of SCC eightfold; in some but not all patient cohorts it raises melanoma >14-fold (see Chapter 238).61 Cancer is now increasing as a result of tanning beds. Individuals whose first sunbed exposure occurred as a young adult, or who had long durations or high frequencies of tanning bed exposure, already have a 70% higher risk of melanoma.62
Characteristics of Ultraviolet Radiation-Induced Cancers and Precancers
In the United States, ∼800,000 BCCs are diagnosed annually, as well as 200,000 SCCs and 70,000 melanomas.11,12 Survival differs strikingly. Fewer than 1 in 10,000 BCCs will metastasize and threaten the patient (see Chapter 115). This number increases to 1 in 40 for SCC, with clinical experience indicating that SCCs on sun-exposed skin are less likely to metastasize than those arising in scars.63 One in seven invasive melanomas is lethal (see Chapter 124). Merkel cell carcinoma (see Chapter 120) is a sun- and polyomavirus-induced cutaneous neuroendocrine cancer that will kill one in three patients diagnosed with it. Its reported incidence has tripled in the past 15 years to approximately 1,500 per year in the United States.64,65
The type of exposure preferentially leading to each malignancy differs. Cumulative lifetime sun exposure is strongly associated with SCC incidence.16,20 BCC and AK instead seem to depend on reaching a certain threshold of UV exposure, often attained in youth, such that sensitive individuals develop BCC at a relatively early age, and the incidence does not increase with further exposure.16,20,66 Case-control studies link melanoma with intense exposure early in life, with one or two blistering sunburns doubling the melanoma risk (see Chapter 124).67
Children are particularly sensitive to sunlight: moving from England to Australia before age 20 years confers the higher Australian incidence of AK, SCC, BCC, and melanoma, but the risk is much less when adults immigrate.68,69 This is not simply due to children spending more time outdoors, as <25% of lifetime exposure occurs before age 18 years.70 One explanation may be that mutant cells created in youth have more years in which to acquire the additional genetic requirements for cancer.
Precursor lesions for SCC (AK) and melanoma (nevi) are also usually related to sun exposure.14,15,71–73 Accounting for roughly 3 million physician visits each year in the United States, AKs (see Chapter 113) are the fourth leading cause for a visit to a dermatologist.74 They typically manifest as 1–3-mm scaly papules that involve erythema but often are easier felt than seen. They proceed to SCC in ∼1% of cases if left untreated.75 Actinic cheilitis is an analogous precancerous state on the sun-exposed lip. The importance of ongoing sun exposure is made apparent by clinical studies indicating that diminishing sun exposure can reduce the number of AKs over a span of months. In randomized sunscreen studies, statistically significant decreases in AKs were seen in the sunscreen group over brief periods.76,77 Common nevi and especially clinically atypical nevi can be precursors of malignant melanoma, with abundant nevi conferring a tenfold risk for cancer.22,78 Acquired melanocytic nevi begin to appear at age 1–5 years in proportion to sunlight exposure and are most frequent in individuals with freckles and red hair.71,72,79 Acquired nevi typically carry the BRAF mutations seen in non-CSD melanomas, but congenital nevi instead have NRAS mutations.80
Ultraviolet Radiation-Induced Genetic Alterations
The first molecular step in sunlight-induced carcinogenesis occurs when UVB photons induce DNA photoproducts (Fig. 112-1). UVB and UVC tend to be absorbed at the 5–6 double bond of pyrimidines (thymine and cytosine), allowing the bond to open.81 If two adjacent pyrimidines are activated, their open bonds cross-react. This creates two single bonds (5–5 and 6–6) that result in a cyclobutane pyrimidine dimer (CPD; eFig. 112-1.1A). The most frequent is TT, but TC, CT, and CC cyclobutane dimers are also made. A single bond between the 6 position of one pyrimidine and the exocyclic group of the other instead creates a pyrimidine (6–4) pyrimidone photoproduct ((6–4)PP) (eFig. 112-1.1A).82 The most frequent (6–4)PP is TC. Both photoproducts distort the DNA helix and are recognized by DNA repair enzymes. (6–4)PPs make the DNA helix more distorted, and thus are recognized and repaired much more rapidly than CPDs.83,84 Half-lives of TT cyclobutane dimers and (6–4)PPs in human skin are 33 hours and 2 hours, respectively.85 UVB induces 500 photolesions per 106 normal bases per J/cm2 in human skin.84
Figure 112-1
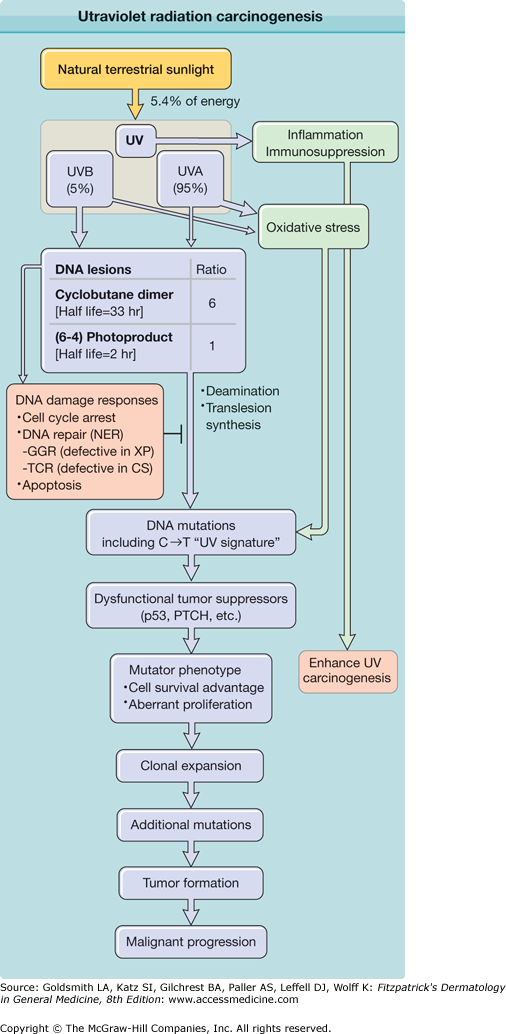
Ultraviolet radiation (UVR) carcinogenesis. UVR induces two major DNA lesions at dipyrimidine sites: cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidone photoproducts [(6–4)PPs]. Ratio given [6 CPDs per 1 (6–4)PP] is that induced by simulated sunlight. UVB accounts for only 5% of UVR (∼0.3% of all terrestrial sunlight energy), but produces the majority of UV-induced DNA lesions. Cells respond to UVR-induced DNA damage by activating DNA damage signaling pathways and inducing cell cycle arrest. Damaged DNA is repaired by nucleotide excision repair (NER), whose subpathways are global genome repair (GGR; defective in xeroderma pigmentosum, XP) and transcription-coupled repair (TCR; defective in Cockayne’s syndrome, CS). Despite their DNA repair deficiency, individuals with CS exhibit no increased incidence of UV-induced skin cancer, likely due to enhanced intracellular accumulation of p53 and apoptosis. (6–4)PPs are rapidly repaired by NER. Unrepaired DNA lesions lead to genetic mutations through deamination or error-prone translesion synthesis. Unrepaired cytosine-containing CPDs contribute to UV signature mutation: C→T transition. Mutations in genes responsible for carcinogenesis confer mutator phenotype including resistance to apoptosis. Clonal expansion of mutant cells increases opportunities to receive additional mutations for tumor development.
eFigure 112-1.1
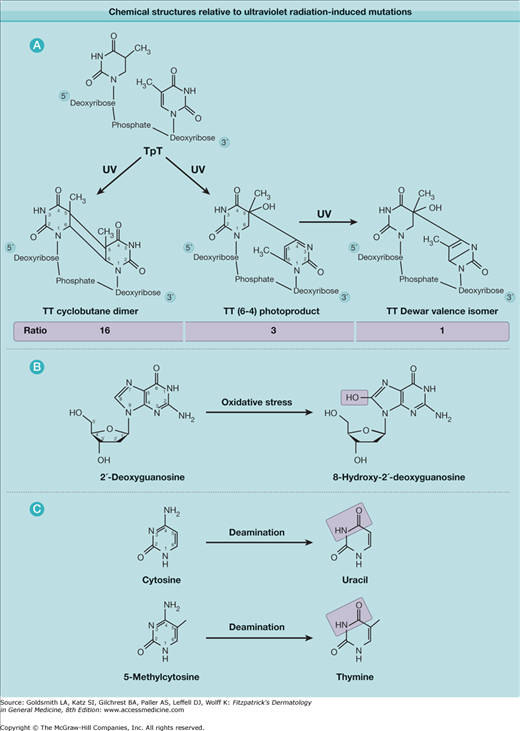
Chemical structures relevant to ultraviolet radiation-induced mutations. A. Thymine dimeric photoproducts. Ratio of amount of DNA lesions induced by simulated sunlight is shown for the three main lesion types. B. Oxidative lesion associated with UVA radiation. C. Deamination that contributes to mutagenesis.
Although there is 20-fold more UVA than UVB in sunlight, UVA requires up to 1,000-fold greater doses for some of its biological effects such as DNA damage.45–47,86–88 Minimal erythemal doses (MEDs) at 300 nm (UVB) and at 360 nm (UVA) for skin type II are 25 mJ/cm2 and 32,000 mJ/cm2, respectively.89 UVA induces T-containing cyclobutane dimers and lesser numbers of oxidized purines and pyrimidines and single-strand breaks.84,90 UVA generates these lesions indirectly by photosensitization (see Chapter 90). UVA also efficiently photoisomerizes UVB-induced (6–4)PPs to their poorly repaired and highly mutagenic Dewar valence isomers88,90 (eFig. 112-1.1A).
 Both UVB and UVA can be absorbed by cytoplasmic ring-containing molecules such as NADH (reduced form of nicotinamide-adenine dinucleotide), riboflavin, quinones, tryptophan and tyrosine, and the heme group of catalase (see Chapter 90). The resulting energetic molecule can interact with DNA to produce a T-containing cyclobutane dimer90 or can produce reactive oxygen species. In the latter pathway, the chromophore’s energy is transferred to oxygen, resulting in singlet oxygen (1O2) or, if an electron is transferred, superoxide (O2•−). In the presence of water, these lead to hydrogen peroxide (H2O2) and thence, in the presence of Fe2+, the hydroxyl radical (•OH). Hydroxyl radicals produce oxidative DNA damage resembling that after γ radiation.91 Reactive oxygen species react with lipid membranes and the redox-sensitive catalytic site of phosphatases (see Section “Cytoplasmic Signaling”). Their production of 8-hydroxy-2′-deoxyguanosine (8-OHdG; eFig. 112-1.1B) in DNA probably accounts for the occasional non-UVR-like mutations after UVB. UVR also upregulates nitric oxide (NO), a more stable radical species that can participate in similar reactions after diffusing long distances and traversing lipid membranes.92
Both UVB and UVA can be absorbed by cytoplasmic ring-containing molecules such as NADH (reduced form of nicotinamide-adenine dinucleotide), riboflavin, quinones, tryptophan and tyrosine, and the heme group of catalase (see Chapter 90). The resulting energetic molecule can interact with DNA to produce a T-containing cyclobutane dimer90 or can produce reactive oxygen species. In the latter pathway, the chromophore’s energy is transferred to oxygen, resulting in singlet oxygen (1O2) or, if an electron is transferred, superoxide (O2•−). In the presence of water, these lead to hydrogen peroxide (H2O2) and thence, in the presence of Fe2+, the hydroxyl radical (•OH). Hydroxyl radicals produce oxidative DNA damage resembling that after γ radiation.91 Reactive oxygen species react with lipid membranes and the redox-sensitive catalytic site of phosphatases (see Section “Cytoplasmic Signaling”). Their production of 8-hydroxy-2′-deoxyguanosine (8-OHdG; eFig. 112-1.1B) in DNA probably accounts for the occasional non-UVR-like mutations after UVB. UVR also upregulates nitric oxide (NO), a more stable radical species that can participate in similar reactions after diffusing long distances and traversing lipid membranes.92
 Nucleotide excision repair (NER; see Chapter 110) is the key protection mechanism against the lethal and mutagenic effects of UVR-induced cyclobutane dimers and (6–4) photoproducts. Two NER pathways have been identified, global genome repair (GGR) and transcription-coupled repair (TCR).93 GGR removes DNA lesions throughout the genome, whereas TCR is specialized for DNA lesions in the transcribed strand of transcriptionally active genes. In humans, excision repair requires the concerted action of six repair factors (XPA, RPA, XPC, TFIIH, XPG, and XPF-ERCC1) composed of nearly 20 polypeptides, some identified and named according to the seven complementation groups of xeroderma pigmentosum (XP). The enzymatic steps of NER include: (1) recognizing damaged DNA, (2) forming dual incisions that bracket the UV lesion, (3) removing the damaged oligomer (24–32 nucleotides in length), (4) gap filling by DNA synthesis, and (5) ligating the repaired strand.94 GGR is considered error-free because the complementary undamaged strand is used as a template for repair synthesis.
Nucleotide excision repair (NER; see Chapter 110) is the key protection mechanism against the lethal and mutagenic effects of UVR-induced cyclobutane dimers and (6–4) photoproducts. Two NER pathways have been identified, global genome repair (GGR) and transcription-coupled repair (TCR).93 GGR removes DNA lesions throughout the genome, whereas TCR is specialized for DNA lesions in the transcribed strand of transcriptionally active genes. In humans, excision repair requires the concerted action of six repair factors (XPA, RPA, XPC, TFIIH, XPG, and XPF-ERCC1) composed of nearly 20 polypeptides, some identified and named according to the seven complementation groups of xeroderma pigmentosum (XP). The enzymatic steps of NER include: (1) recognizing damaged DNA, (2) forming dual incisions that bracket the UV lesion, (3) removing the damaged oligomer (24–32 nucleotides in length), (4) gap filling by DNA synthesis, and (5) ligating the repaired strand.94 GGR is considered error-free because the complementary undamaged strand is used as a template for repair synthesis.
 This core machinery of GGR is also used by TCR in active genes. Whereas in GGR the XPC protein recognizes distortions in the DNA double helix, the damage recognition signal for TCR is an RNA polymerase II complex stalled at a UV lesion, which attracts the core GGR machinery. RNA polymerase II sterically hinders the accessibility of NER factors and is therefore removed from the damage site by the CSA and CSB proteins. CSA and CSB are the genes mutated in Cockayne’s syndrome, an autosomal recessive disorder characterized by cutaneous photosensitivity and physical and mental retardation.95 Induction of these repair factors is genetically regulated (see Section “DNA Damage Signaling”).
This core machinery of GGR is also used by TCR in active genes. Whereas in GGR the XPC protein recognizes distortions in the DNA double helix, the damage recognition signal for TCR is an RNA polymerase II complex stalled at a UV lesion, which attracts the core GGR machinery. RNA polymerase II sterically hinders the accessibility of NER factors and is therefore removed from the damage site by the CSA and CSB proteins. CSA and CSB are the genes mutated in Cockayne’s syndrome, an autosomal recessive disorder characterized by cutaneous photosensitivity and physical and mental retardation.95 Induction of these repair factors is genetically regulated (see Section “DNA Damage Signaling”).
UVR-damaged DNA contains characteristic “signature” mutations that are readily detected decades after sun exposure. These mutations have been used to answer many questions about the origin of cancer. A CPD can lead to a mutation in two ways (Fig. 112-2). When the lesion is copied during DNA replication, the DNA polymerase may read a damaged cytosine as a thymine and insert an adenine opposite it. At the next round of replication, the polymerase correctly inserts thymine across from adenine, with the result being a C→T substitution. Although the TT cylobutane dimer is the best known and most frequent photoproduct, the thymines are not mutagenic because the XPV gene encodes a specialized polymerase (pol eta) that adds adenines across from a T-containing cyclobutane dimer.96,97 Alternatively, a mutation can arise because cyclobutane dimers accelerate spontaneous deamination of their cytosines to uracil (eFig. 112-1.1C), leading to a C→T substitution; no polymerase error is involved.98 Deamination of cytosines to uracil within photolesions is increased at the transcribed strand, presumably mediated by persistent stalling of the transcription complex at the photolesion.99 In either case, C→T mutations occur only where a cytosine lies next to a thymine or another cytosine, because the major UV photoproducts join adjacent pyrimidines. If two adjacent cytosines mutate, the result is CC→TT. This distinctive pattern of mutation, C→T where the C lies next to another pyrimidine, including CC→TT, is unique to UVR and is called the UV signature mutation.100
Figure 112-2
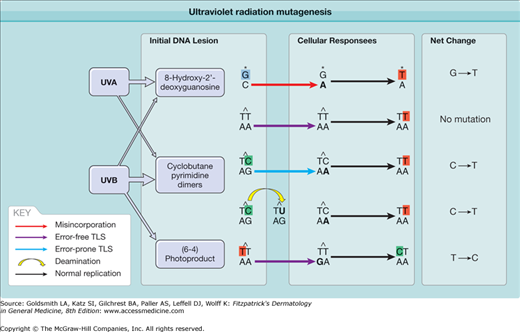
Ultraviolet radiation mutagenesis: how replication through unrepaired DNA lesions leads to DNA mutation. Cyclobutane pyrimidine dimers (CPDs) are the most relevant DNA lesions for UV mutagenesis and are primarily induced by UVB. Thymine cyclobutane dimers are most abundant but not mutagenic, because error-free translesion synthesis (TLS) correctly adds adenines opposite a thymine dimer. Cytosines within CPDs are mutagenic: error-prone TLS mistakenly adds adenine opposite cytosine, or deamination converts cytosine to uracil. This leads to the UV signature mutation, a C→T transition. The (6–4) photoproduct induced by UVB and 8-hydroxy-2′-deoxyguanosine induced by oxidative stress via UVA are also mutagenic but relatively rare.
UV signature mutations (Table 112-1) provide a tool for deducing backward from mutations found in tumors to the original carcinogen. Nearly all experimentally created UVB or UVC mutations are located at adjacent pyrimidines, and about two-thirds are signature mutations.100 The remaining third, typically G→T and T→C substitutions or one to two base insertions or deletions, are still caused by UV but probably arise indirectly by photosensitization-produced reactive oxygen species. G→T transversion can be caused by incorporation of adenine opposite 8-hydroxy-2′-deoxyguanosine (8-OHdG; also called 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG)) (Fig. 112-2),101,102 a common oxidative DNA lesion that remains at a high level in the epidermis even 7 days after UVR exposure.103 Because this oxidative class of damage can be caused by many carcinogens, these mutations do not reveal whether their source was UVB, UVA, tobacco smoke, or intracellular oxidative phosphorylation. However, tumors carrying classic UV signature mutations must also contain UV-induced oxidative mutations. UVA, in contrast, only weakly induces UVB signature mutations by photosensitization but also generates oxidation-like mutations and T→G changes. The latter are rare with UVB or other carcinogens and have been proposed to be a UVA fingerprint,104,105 although the mechanism of induction of T→G mutations remains unclear.
Classical UVB signature mutations | C→T (CC→TT) at dipyrimidine sites |
Oxidative mutations (could be induced by UVA) | G→T |
UVA signature mutations | T→G |
PUVA-type mutations | Mutations at the T of TA, TG, or TT sites |
Although (6–4)PPs are rapidly repaired, they are mutagenic if they remain unrepaired. Error-prone DNA polymerases add a guanine opposite a thymine of (6–4)PP, resulting in a T→C mutation (Fig. 112-2).106
UV signature mutations identified p53 as critical for preventing SCC and BCC but not melanoma.100,107 The p53 protein is a transcription factor that controls genes involved in the cell cycle, apoptosis, and DNA repair; it also acts directly on apoptosis proteins.7 The p53 gene is mutated in about one-half of all human cancers and is termed a tumor suppressor gene because cancer arises from losing its normal function rather than gaining an abnormal function as for oncogenes. Over 90% of SCC in US patients contain these mutations, as well as three-fourths of AK.108 Although nearly all BCCs overexpress p53 protein, only half carry p53 mutations. Each mutation changes the amino acid, indicating that the mutation was selected for and contributed to tumor development, rather than being simply an indicator of sun exposure. These p53 mutations are most frequent at nine mutation hot spots in important functional regions of the protein. Compared to internal cancers, some skin cancer hot spots are displaced several nucleotides to lie at a site of adjacent pyrimidines.107 Some sites may be hot spots because repair is slower there.109 Other skin hot spots, like internal cancer hot spots, lie at 5-methylCG sites where body temperature slowly deaminates 5-methylcytosine to thymine; UVR accelerates this process.
The p53 mutations in AK indicate that these dysplasias are clonal; patients with multiple AKs have different mutations in discrete skin lesions.108,110,111 The similarity of AK and SCC mutations supports the idea that AKs can progress to SCC. SCCs have more oxidative mutations than AKs, implying the involvement of oxidative stress in progression of AK toward SCC.105 Aggressive skin tumors from patients exposed to both sun and chemicals contain multiple distinct p53 mutation patterns, as if several tumors arose in an abnormal field and intermingled.112 XP tumors contain very frequent CC→TT mutations, perhaps because slow repair allows more time for cytosine deamination.113,114 Double-base mutations are also seen in conjunctival SCC, a tumor associated with human immunodeficiency virus (HIV) in sunny areas.115 Sunscreens reduce the level of UV signature mutations.116 In contrast, arsenic-induced BCC and SCC have non-UV p53 mutations; p53 mutations in BCCs from sun-shielded body sites resemble those seen with oxidative damage.117,118 UV signature mutations in p53 can also be seen in Merkel cell carcinoma.119
Sunlight mutates p53 early.108,120 Normal sun-exposed skin carries ∼60,000 clones of p53-mutant keratinocytes, three to 3,000 cells in size.121 By hematoxylin and eosin staining, the cells in these mutant clones appear completely normal.44 The early appearance of p53 mutations makes it possible to trace lineages in tumor development. Microdissecting lesions containing AK, carcinoma in situ, and SCC reveals that each stage contains the same p53 mutation.122 Although this result shows that each stage arose from the same founder lesion, it does not show that the stages derived from each other. To show a lineage, it is necessary to find additional mutations that appear in succession. In microdissected BCCs, one p53 mutation is present throughout the tumor, with various second mutations in different regions of the tumor.123 Once both p53 alleles are mutated, the cell is prone to aneuploidy,124,125 increasing the likelihood of a mutator phenotype.
Most sporadic BCCs have inactivating mutations in the PTCH tumor suppressor gene, a part of the hedgehog pathway (see Chapter 115); the remainder has activating mutations in its target, SMO. The hedgehog pathway appears to be a “gatekeeper” for basal cell carcinogenesis, needing to be mutated early in BCC: minute BCCs have PTCH mutations, as do all histological subtypes; no BCCs have loss on other chromosomes without involvement of PTCH; and a congenital lesion that can progress to BCC, the sebaceous nevus, has PTCH allelic loss in 40% of cases.126–128
In sporadic BCCs, about three-fourths of PTCH mutations are UVR-like (either UV signature mutations or the expected UVR-induced oxidative mutations) and a further 15% are 1- or 2-base insertions or deletions, often adjacent to a C→T at a dipyrimidine site.129 BCCs from XP patients contain UVR-like PTCH and SMO mutations, with CC→TT mutations overrepresented.130,131 About 20% of the mutations in sporadic BCCs are not UVR-like and resemble germline mutations seen in basal cell nevus syndrome patients—deletions or insertions larger than two base pairs. This finding may relate to the clinical observation that one-third of BCCs occur on parts of the body that are not chronically sun-exposed, as well as the correlation between truncal BCC and defects in the glutathione radical-scavenging system (see Section “Genetic Risk Factors for Ultraviolet Radiation Carcinogenesis”). PTCH mutations tend to code for stop codons or frameshifts that completely inactivate the protein. In hereditary BCCs, nearly all tumors arose after losing the normal allele. This allelic loss appears related to sunlight, as NBCCS tumors are most frequent on sun-exposed skin and are rare in blacks.39 UVB rarely causes this type of large genetic rearrangement so, in analogy to X-ray sensitivity of NBCCS patients, UVA-photosensitized reactive oxygen may be important.
(See Chapter 238.)
In psoralen-treated cells, UVA forms adducts at TA, TG, or TT sequences, as well as cross-links between the two DNA strands at these sites. In human PUVA-induced keratoses, SCCs, and BCCs, about one-fourth of mutations in p53 or HRAS are psoralen-like mutations at the T of TA, TG, or TT; this proportion increases as the PUVA dose increases.132,133 However, the majority of mutations are UV signature mutations. The UVR-like mutations could have arisen from the UVA in PUVA, UVB treatment for psoriasis, or environmental UVB.
(See Chapter 124.)
Despite the correlation between melanoma and sunlight and the fact that UVB signature mutations are present in melanoma,134,135 genes with UV signature mutations for melanoma development are not prevalent. The CDKN2A locus is frequently mutated or deleted in familial and sporadic melanoma. Its two distinct tumor suppressor proteins, INK4A (also known as p16) and ARF, inhibit cell cycle progression via Rb and p53, respectively. INK4A inhibits CDK4/6’s inactivation of the retinoblastoma protein, Rb. ARF inhibits MDM2-mediated degradation of p53. Germline mutations in CDKN2A are observed in ∼25%–40% of familial melanomas.136,137 In sporadic melanoma, allelic loss of CDKN2A is more common than the rare INK4A inactivating mutations138–140 or inactivation of INK4A by promoter methylation.141 The role of sunlight in these genetic events is unknown.
Oncogene mutations are found in the RAS-BRAF-MEK-ERK mitogen-activated protein kinase (MAPK) signaling pathway. This signaling cascade is normally activated on growth factor stimulation, and its sequential phosphorylation regulates cell proliferation and differentiation. Activating mutations in MAPK signaling remove the growth factor requirement. RAS mutations are present in 10%–20% of melanomas and have been correlated with UVR exposure.142–144 The most prevalent RAF mutation in melanomas, the BRAF V600E point mutation, renders the kinase constitutively active and enhances ERK activation.142 This mutation is present in ∼80% of acquired melanocytic nevi, suggesting a potential early role of BRAF in melanoma development.144 The V600E mutation is not UVR-like and is associated with intermittent sunlight exposure.27,29
Ultraviolet Radiation-Induced Steps in Cancer and Cancer Prevention
Skin tumors arise on a background of sun-damaged skin. To prevent sun damage, the skin reacts to acute and chronic UVR exposure by multiple stress responses.
 (See Chapter 110)
(See Chapter 110)
 A cell with damaged DNA upregulates normal p53 protein.145,146 UV signaling is initiated by cyclobutane dimers and (6–4) photoproducts specifically in the small minority of actively transcribed genes, with stalled RNA polymerase both recruiting excision repair proteins and initiating signaling.6,7 In an unknown way, this activates ATR and CHK1 kinases, which phosphorylate p53 at sites that make it resistant to proteasomal degradation mediated by HDM2 (MDM2 in mouse). p53 then transcriptionally activates a large repertoire of genes, including the repair proteins p48, which is required for GGR and is defective in XP group E, and GADD45. Additionally, p53 functions as a chromatin accessibility factor, modifying the structure of damaged DNA and making it more accessible to repair factors. p53 also transactivates the cell cycle arrest protein p21, although in keratinocytes UVR induces p21 even without p53.147 UVR primarily slows down S phase (“S phase delay”) and induces a modest G2 arrest, unlike ionizing radiation, which uses p53 to induce G1 arrest. It is often said that cell cycle arrest facilitates DNA repair and survival, but there is little evidence supporting this concept; deleting the p21 cell cycle arrest protein has no effect on repair after UVR exposure.148 These “guardian of the genome” roles of p53 are complemented by a “cellular proofreading” role in which p53 erases aberrant cells by apoptosis rather than repairing them: it transcriptionally activates the death receptor Fas and proapoptotic effectors Bax (Bcl-2–associated X protein), Bak, Bid, and PUMA; it directly activates Bax protein at the mitochondrion; and it inactivates E2F1, which otherwise inhibits antiapoptotic Bcl-2.8 This suite of UV responses is lost when p53 is mutated by sunlight. DNA damage also activates the cytoplasmically sequestered transcription factor nuclear factor κB (NFκB), which then activates proinflammatory cytokines such as interleukin 10, growth signals, and antiapoptotic signals.149,150 Finally, DNA photoproducts trigger UVR-induced systemic immunosuppression and suppression of dendritic cell antigen-presenting activity, but do not contribute to inflammatory edema.151,152
A cell with damaged DNA upregulates normal p53 protein.145,146 UV signaling is initiated by cyclobutane dimers and (6–4) photoproducts specifically in the small minority of actively transcribed genes, with stalled RNA polymerase both recruiting excision repair proteins and initiating signaling.6,7 In an unknown way, this activates ATR and CHK1 kinases, which phosphorylate p53 at sites that make it resistant to proteasomal degradation mediated by HDM2 (MDM2 in mouse). p53 then transcriptionally activates a large repertoire of genes, including the repair proteins p48, which is required for GGR and is defective in XP group E, and GADD45. Additionally, p53 functions as a chromatin accessibility factor, modifying the structure of damaged DNA and making it more accessible to repair factors. p53 also transactivates the cell cycle arrest protein p21, although in keratinocytes UVR induces p21 even without p53.147 UVR primarily slows down S phase (“S phase delay”) and induces a modest G2 arrest, unlike ionizing radiation, which uses p53 to induce G1 arrest. It is often said that cell cycle arrest facilitates DNA repair and survival, but there is little evidence supporting this concept; deleting the p21 cell cycle arrest protein has no effect on repair after UVR exposure.148 These “guardian of the genome” roles of p53 are complemented by a “cellular proofreading” role in which p53 erases aberrant cells by apoptosis rather than repairing them: it transcriptionally activates the death receptor Fas and proapoptotic effectors Bax (Bcl-2–associated X protein), Bak, Bid, and PUMA; it directly activates Bax protein at the mitochondrion; and it inactivates E2F1, which otherwise inhibits antiapoptotic Bcl-2.8 This suite of UV responses is lost when p53 is mutated by sunlight. DNA damage also activates the cytoplasmically sequestered transcription factor nuclear factor κB (NFκB), which then activates proinflammatory cytokines such as interleukin 10, growth signals, and antiapoptotic signals.149,150 Finally, DNA photoproducts trigger UVR-induced systemic immunosuppression and suppression of dendritic cell antigen-presenting activity, but do not contribute to inflammatory edema.151,152
 “The UV response” initially referred to the p53-independent activation of JNK, its target c-JUN, and, via the FOS-JUN transcription factor AP-1, induction of genes for collagenase, metallothionein, and c-JUN and c-FOS themselves.153 The signal begins when reactive oxygen species generated by UVB or UVA photosensitization inactivate phosphatases by converting a highly sensitive cysteine residue in the catalytic site to sulfenic acid.154,155 Dephosphorylation of growth factor receptor dimers and death receptor trimers is slowed, leading within minutes to more phosphorylated active receptors. Activated death receptors (involved in apoptosis) such as Fas and tumor necrosis factor-α (TNF-α) receptor then cluster even without a ligand, recruiting the adapter proteins DAXX and FADD, and activating cytoplasmic kinases and scaffold proteins.156–161 These activate ASK1 and MEKK4/7 kinases and their target, JNK.162 Phosphatase inhibition can activate JNK directly. In parallel, AP-1 also induces genes for the death receptor ligands, FasL and TNF-α.163–165 These ligands, together with UV upregulation of FAS receptor via p53,166 create a delayed feedback loop that, as with ionizing radiation,167 appears to prolong the rapid but transient response triggered by UVR’s inactivation of phosphatases. Without this prolongation, UVR-activated NFκB quickly terminates the JNK response.168,169 This loop is important because transient JNK activation leads to cell proliferation, but constitutive JNK activation induces apoptosis.170,171 AP-1 also induces immunomodulatory cytokines such as interleukin 12, which facilitates NER172; the AP-1-induced metalloproteinases degrade dermal extracellular matrix molecules, such as collagen, and may contribute to photoaging.173 UVR also blocks initiation of protein translation, via kinases that inactivate elongation factor eIF2α.174
“The UV response” initially referred to the p53-independent activation of JNK, its target c-JUN, and, via the FOS-JUN transcription factor AP-1, induction of genes for collagenase, metallothionein, and c-JUN and c-FOS themselves.153 The signal begins when reactive oxygen species generated by UVB or UVA photosensitization inactivate phosphatases by converting a highly sensitive cysteine residue in the catalytic site to sulfenic acid.154,155 Dephosphorylation of growth factor receptor dimers and death receptor trimers is slowed, leading within minutes to more phosphorylated active receptors. Activated death receptors (involved in apoptosis) such as Fas and tumor necrosis factor-α (TNF-α) receptor then cluster even without a ligand, recruiting the adapter proteins DAXX and FADD, and activating cytoplasmic kinases and scaffold proteins.156–161 These activate ASK1 and MEKK4/7 kinases and their target, JNK.162 Phosphatase inhibition can activate JNK directly. In parallel, AP-1 also induces genes for the death receptor ligands, FasL and TNF-α.163–165 These ligands, together with UV upregulation of FAS receptor via p53,166 create a delayed feedback loop that, as with ionizing radiation,167 appears to prolong the rapid but transient response triggered by UVR’s inactivation of phosphatases. Without this prolongation, UVR-activated NFκB quickly terminates the JNK response.168,169 This loop is important because transient JNK activation leads to cell proliferation, but constitutive JNK activation induces apoptosis.170,171 AP-1 also induces immunomodulatory cytokines such as interleukin 12, which facilitates NER172; the AP-1-induced metalloproteinases degrade dermal extracellular matrix molecules, such as collagen, and may contribute to photoaging.173 UVR also blocks initiation of protein translation, via kinases that inactivate elongation factor eIF2α.174
UV signaling generates sunburn cells—basal and suprabasal keratinocytes with dense, pyknotic nuclei and intensely eosinophilic cytoplasm.175 This apoptotic morphology is accompanied by pathognomonic DNA double-strand breaks and cleaved caspase 3. UVR-induced apoptosis requires signals from both DNA damage and the cytoplasm: DNA photoproducts in active genes trigger p53 and its regulator MDM2,108,176 but apoptosis also requires JNK and is partially blocked by antioxidants.177,178 Although TNF-α is required, injecting TNF-α does not lead to sunburn cells, so UV-induced cytoplasmic signaling is not sufficient.179,180 In fibroblasts, keratinocytes, or melanocytes with normal p53 and irradiated with physiologic UVB or UVC doses, apoptosis proceeds through the intrinsic mitochondrial pathway rather than the death-receptor/caspase 8 pathway.178,181
Apoptosis then prevents cancer by removing UVR-damaged cells in a process termed “cellular proofreading”.182 Mice accumulate mutations at a rapid rate if they are defective in apoptosis due to a defect in p53 or Fas ligand, or due to overexpressing the antiapoptotic protein Survivin.165,183,184 In Survivin’s case, this increases SCC.184 The epidermal hyperplasia that occurs several days after UVR exposure may replace cells lost by apoptosis or may remove additional damaged or mutant cells by desquamation. The signal for hyperplasia involves both DNA photoproducts and the epidermal growth factor receptor.185 UVA inhibits UVB-induced apoptosis, suggesting that UVA may enhance UV carcinogenesis by disrupting elimination of UV-damaged cells.186
Dermal fibroblasts have a critical role in maintaining appropriate UVR responses of epidermal keratinocytes. The insulin-like growth factor 1 (IGF-1) secreted by normal human fibroblasts is required for appropriate induction of senescence of keratinocytes after UVR. UV-induced senescence protects keratinocytes from propagating UVR-induced mutations. In human geriatric skin, expression of IGF-1 is decreased in dermis and the aged keratinocytes proliferate in the presence of UV-damaged DNA, possibly leading to an increased carcinogenic potential in the skin of older individuals.187
In chronically UVB-exposed human skin, p53-mutant clones are found at both sites of epidermal stem cells (see Chapter 45): the hair follicle, whose bulge region contributes to follicle development and transiently to wound repair, and the interfollicular epidermis, which maintains epidermal homeostasis and can also generate follicles.121,188,189 SCCs are thought to originate in interfollicular epidermis, whereas histologic evidence and the expression pattern of PTCH indicate that BCCs originate in the follicles.190,191 Hedgehog signaling through PTCH is crucial for maintaining skin stem cell populations and for regulating hair follicle and sebaceous gland development.192 Chronically UV-irradiated human or mouse skin contains scattered basal cells with unusually high levels of DNA photoproducts.193 The tumor promoter TPA (12-O-tetradecanoylphorbol 13-acetate), which induces skin stem cells to proliferate,194 causes these cells to disappear and be replaced by clusters of p53-mutant keratinocytes. This behavior resembles that of stem cells that are quiescent and poorly repaired, at least on the parental DNA strand, until triggered to divide.195 The immortalization-promoting enzyme telomerase is normally present only in the epidermal basal layer, but is elevated in sun-exposed skin, skin precancers, and cancers.196,197
A single mutant cell must clonally expand to reach a clinically discernible size. Less obviously, clonal expansion facilitates the multiple genetic hit mechanism of cancer. Physiologic UVR doses create mutations at a frequency of ∼10−4/gene per cell division.198 The specific mutations needed to activate an oncogene are infrequent. Spontaneous mutations, which reflect errors by the replication machinery or DNA damage due to body temperature, are also rare, on the order of 10−5/gene per cell division.198 The probability of mutating five genes, such as an oncogene and both alleles of two particular tumor suppressor genes, is then at best 10−20. Accounting for the 60% lifetime expectation of skin cancer in Australia solely in terms of simultaneous genetic hits in one cell is impossible. In contrast, clonal expansion increases by 1,000-fold the number of targets for the next mutation.
A stem cell’s clonal expansion is normally limited to its stem cell compartment.199 Sunlight is a key driver of clonal expansion beyond this point. The p53-mutant clones in human skin are larger in chronically sun-exposed skin.121 In mice, p53-mutant clones stop growing and regress when UVB treatment ends, indicating that clone expansion is due to an ongoing UVR-induced physiologic event rather than an irreversible mutation.199,200 One of these physiologic events is UVR-induced apoptosis.184 Once a p53 mutation arises, the cellular proofreading mechanism backfires: subsequent UVR exposures eliminate damaged normal cells but spare apoptosis-resistant mutants. An apoptosis-resistant mutant is no longer restrained within its stem cell compartment and escapes to colonize the adjacent apoptosis-sensitive compartment. Repeating this process further enhances clonal expansion. AKs also often regress once irradiation stops,201,202 but SCCs do not, indicating that invasive tumors no longer need a promoter. Apoptosis thus has diverse effects on the stages of skin cancer progression: it prevents new p53 mutations from initially arising, facilitates the expansion of p53-mutant clones and papillomas (via death of apoptosis-capable cells adjacent to mutant clones), and suppresses the mutations that convert a papilloma to SCC.165,184
Cell–cell interactions prevent abnormal cells from proliferating inappropriately. In human autotransplant experiments, BCCs transferred from their original site regressed—suggesting that an abnormal underlying dermis is required for the tumor to persist.203 Dermal fibroblasts suppress transformed keratinocytes by secreting transforming growth factor-β that induces squamous differentiation.204 Normal keratinocytes also suppress their transformed neighbors, and UVR interferes with these signals. Normal human keratinocytes eliminate adjacent transformed keratinocytes (mutated in p53 and HRAS) by inducing cell cycle arrest and differentiation.205 Physiologic doses of UVB cause apoptosis and differentiation in the normal cells, but not in the transformed keratinocytes, allowing the latter to clonally expand.206 Other intercellular signals are mediated by integrins—membrane receptors for extracellular matrix proteins such as collagen (α2β1 integrin), laminin (α3β1), and fibronectin (α5β1). In keratinocytes, integrins bound to such ligand provide a “do not differentiate” signal through MAPK pathways, suppressing keratinocyte apoptosis, and allowing a stem cell pool to be maintained.207,208 Integrin receptors are often dysregulated in tumors. UVB irradiation downregulates the β1 integrin subunit. UVA downregulates the gap junction communication protein connexin 43, resembling the action of the tumor promoter TPA.209,210
Melanocyte proliferation is normally regulated by keratinocytes via cell–cell adhesion receptors such as E-cadherin, P-cadherin, and desmogleins; these receptors are lost in vertical growth phase melanomas.211 UVR stimulates keratinocytes to secrete endothelin-1, which then downregulates melanocyte E-cadherin and upregulates N-cadherin.212 This E- to N-cadherin switch diverts melanocyte interactions away from keratinocytes and toward fibroblasts and melanocytes.211 Endothelin also downregulates the α6 integrin subunit,213 upregulates the αvβ3 and α2β1 integrins that anchor melanocytes to dermal collagen and are associated with vertical growth phase melanoma, inhibits gap junction communication by phosphorylating connexin 43, and activates metalloproteinases associated with basement membrane invasion.
In humans, the primary evidence cited for immune surveillance in preventing UVR-induced skin cancer is the 10–20-fold increase in AK and SCC on previously sun-exposed skin in transplant patients receiving chronic immune suppression to prevent organ rejection. In one Australian study, 27% of deaths in a heart transplant cohort were due to skin cancer.214 The increase begins months to a few years after immune suppression is initiated, and both the AKs and SCCs are unusually aggressive.215–219 Melanomas are also increased.220 Several lines of evidence complicate the data that support a role for immune surveillance. Cyclosporine promotes tumor growth in vitro and in immune-deficient mice in which there is no immune system to be suppressed.221 Azathioprine, often a component of organ rejection therapy, is a mutagen when followed by UVA irradiation.222 It may thus not only be working via immune suppression. HIV patients do have a modestly increased incidence of SCC, but these tumors at sun-shielded sites are associated with human papillomavirus (HPV).223 Skin cancers are often said to develop in patients who are immunodeficient due to leukemia or lymphoma. Complicating interpretation, these patients have often received cyclophosphamide, a known mutagen. Most published reports lack controls or patient data, but an eightfold elevation in chronic lymphocytic leukemia seems valid.224,225
Merkel cell carcinoma, in part caused by a virus and by UV, appears to be truly sensitive to immune function. Its incidence increases tenfold not only in solid-organ transplant recipients but also in HIV patients (∼13-fold) and in chronic lymphocytic leukemia (over 30-fold increase) (see Chapter 120). The chance of metastasis and mortality also increases in all types of immunosuppressed patients. Regardless of why immunosuppressed patients are at increased risk of skin cancer, it is critical that they be monitored closely because, when caught early, even tumors in immunosuppressed patients are curable.
Animal Models of Skin Cancer: Clinical Implications
 Showing causality requires manipulating an experimental system, which usually cannot be done in humans. Early experiments generated fibrosarcomas by irradiating mouse ears. SCCs can be generated by irradiating back skin daily for about 4 months with doses of UVB severalfold above the minimal erythemal dose. The sequence of events resembles that in humans, with p53-mutant clones appearing early, followed by reddish lesions that resemble AKs both visibly and histologically.226,227 SCCs induced by UVB contain UV signature p53 mutations.228 p53-mutant clones may be a precursor lesion for SCC.229,230 Growth of an existing SCC no longer depends on UVR.227,231
Showing causality requires manipulating an experimental system, which usually cannot be done in humans. Early experiments generated fibrosarcomas by irradiating mouse ears. SCCs can be generated by irradiating back skin daily for about 4 months with doses of UVB severalfold above the minimal erythemal dose. The sequence of events resembles that in humans, with p53-mutant clones appearing early, followed by reddish lesions that resemble AKs both visibly and histologically.226,227 SCCs induced by UVB contain UV signature p53 mutations.228 p53-mutant clones may be a precursor lesion for SCC.229,230 Growth of an existing SCC no longer depends on UVR.227,231