15 Tumors of the facial skeleton
Fibrous dysplasia
Synopsis
 Craniofacial tumors comprise a wide variety of different tumor types at various locations in the craniofacial region. Management of craniofacial tumors in general is exemplified by the management of craniofacial fibrous dysplasia, discussed in this chapter.
Craniofacial tumors comprise a wide variety of different tumor types at various locations in the craniofacial region. Management of craniofacial tumors in general is exemplified by the management of craniofacial fibrous dysplasia, discussed in this chapter.


 Rapid enlargement and pain may indicate cystic degeneration or malignant transformation.
Rapid enlargement and pain may indicate cystic degeneration or malignant transformation.






Historical perspective
In 1891, while describing hyperparathyroidism, von Recklinghausen inadvertently encountered a pathological condition characterized by fibrotic change in bone, which he termed “osteitis fibrosa generalisata.”1,2 In 1942 Lichtenstein and Jaffe introduced the term “fibrous dysplasia” for a similar condition and further divided the entity into monostotic and polyostotic forms.3 McCune, in 1936, described a patient who presented with the polyostotic form of fibrous dysplasia, precocious puberty, multiple skin pigmentation, and hyperthyroidism.4 Albright et al. described a case with similar presentations in 1937.5 This condition was later referred to as McCune–Albright syndrome.
Basic science/disease process
Fibrous dysplasia is a benign bone lesion in which normal bone is replaced by fibro-osseous tissue. The lesion is reported to represent 5–7% of all benign bone tumors.6 Fibrous dysplasia can present in a single bone (monostotic) or multiple bones (polyostotic), and also can be associated with other conditions such as in McCune–Albright syndrome.
The lesions of fibrous dysplasia develop during skeletal growth and have a variable natural evolution. Clinical presentation may occur at any age, with the majority of the lesions being detected by the age of 30 years. The lesions usually present at around 10 years of age and then progress throughout adolescence. At the initial stage, the lesions commonly present as painless swellings. The lesion may exhibit periods of dormancy intermixed with periods of growth. In many cases the progression of the disease stops after adolescence. However there are cases where the lesions continue to progress after puberty.7 The disease does not appear to have any gender predilection, except in McCune–Albright syndrome, which is more commonly found in females.8
Monostotic fibrous dysplasia is much more common than its polyostotic counterpart, accounting for as many as 80% of cases.8 Although any bone in the body may be affected, the most frequently involved sites are the ribs, long bones, pelvis, jaws, and skull. Most of the lesions affecting the jaws are monostotic disease. On the other hand, skull involvement occurs in 27% of monostotic patients and up to 50% of polyostotic patients.7 In the craniofacial region, fibrous dysplasia frequently involves, in descending order, the maxilla, mandible, frontal bones, sphenoidal bones, ethmoidal bones, parietal bones, temporal bones, and occipital bones.2 Maxillary lesions may extend to include the zygoma, sphenoid bone, maxillary sinus, and floor of the orbit. In the mandible, the body area is the most frequently affected.
Multiple brownish skin pigmentations and endocrinopathies, such as precocious sexual development, hyperthyroidism, or hyperparathyroidism, indicate McCune–Albright syndrome. In McCune–Albright syndrome, the borders of the pigmented skin lesions are typically serrated or irregular, as opposed to the regular margins observed in the café-au-lait spots of patients affected with neurofibromatosis.9 The association of soft-tissue myxoma, usually intramuscular, adjacent to lesions of fibrous dysplasia has been described in Mazabraud syndrome. The term “leontiasis ossea” describes a rare form of polyostotic fibrous dysplasia that involves the frontal and facial bones, resulting in marked deformities resembling a lion’s face.7 Another craniofacial entity, cherubism, is a hereditary fibro-osseous lesion, which symmetrically involves the mandible and maxilla. Although sometimes classified as a variant of fibrous dysplasia, cherubism is likely to be a form of giant cell reparative granuloma.10
Based on histological features, three main types of fibrous dysplasia can be identified: (1) Chinese writing type; (2) pagetoid type; and (3) hypercellular type.2 Each type is differentiated on the basis of the amount, architecture, and cellularity of the bony tissue. The Chinese writing type is commonly found in the long bones and axial skeleton (rib, vertebrae). The bone trabeculae are thin and disconnected, with active osseous resorption by osteoclasts. Frequently resorption of the interior of bone trabeculae is observed (so-called dissecting resorption), similar to those observed in hyperparathyroidism. The osteogenic cells are star-shaped and numerous Sharpey fibers are present. The pagetoid type is commonly found in cases of fibrous dysplasia involving nongnathic craniofacial bones. The appearance is similar to osseous tissue found in Paget’s disease, with dense and sclerotic trabecular tissue. The hypercellular type is characterized by the presence of discontinuous bone trabeculae distributed in an ordered and sometimes parallel fashion. Typically the sides of trabeculae are associated with multiple osteoblasts which are arranged in multiple layers. This type appears to occur commonly in gnathic bones. Like the pagetoid variant, a significant amount of bone is present in the hypercellular type. It has been noted that fibrous dysplasia involving craniofacial bones produces radiologically hyperdense lesions compared to fibrous dysplasia in other sites.11
Many authors accept the premise that fibrous dysplasia represents a nonneoplastic, hamartomatous growth resulting from altered bone cell activity. Studies have shown that somatic mutation of the Gs-alpha gene occurs in osteoblastic cells derived from bone lesion in patients with fibrous dysplasia.12 The mutation was first noted in patients with McCune–Albright syndrome but later was also identified in patients with isolated monostotic and polyostotic fibrous dysplasia. The mutation may induce abnormal osteoblastic cell proliferation or cell function in this disorder. Mutation in the Gs-alpha gene in osteoblastic cells leads to activation of adenylate cyclase, elevated cyclic adenosine monophosphate (cAMP) level and increased proliferation of abnormal osteoblastic cells, which results in overproduction of a disorganized collagenous matrix. Incidentally, it has been noted that an overactive cAMP signaling pathway also stimulates the growth of certain tissues such as gonads, thyroid, adrenal cortex, and melanocytes, leading to endocrinopathies and skin pigmentation, as noted in patients with McCune–Albright syndrome.13 Yamamoto et al. noted that elevated level of cAMP in fibrous dysplasia lesions leads to increased level of interleukin-6 in patients affected by McCune–Albright syndrome. Increased interleukin-6 level in turn stimulates the activation of osteoclasts, leading to bone resorption, which is often observed in bone lesions of fibrous dysplasia.14 This might explain why pamidronate, a bisphosphonate which inhibits osteoclast activity, can potentially produce an increase in the bony density in fibrous dysplasia and delay the spread of the lesions into surrounding bones.15
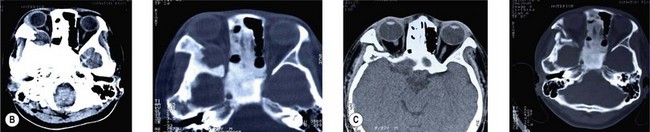
Malignant degeneration occurring within a fibrous dysplasia lesion is relatively uncommon. The malignant lesions usually develop in the third or fourth decade of life.16 Malignancies can occur in both monostotic and polyostotic fibrous dysplasia, with frequency ranges from 0.5% (in monostotic) to 4% in McCune–Albright syndrome.17,18 The most common histotypes are osteosarcoma, followed by fibrosarcoma, chondrosarcoma, and malignant fibrous histiocytoma.19 The common symptoms of sarcomatous change are swelling and pain, usually developing rapidly. Radiographic imaging shows areas with poorly defined margins and osteolytic destruction. The lesions may extend through the bone cortex into the surrounding soft tissue. Periosteal reaction is usually not prominent. A coexisting aneurysmal bone cyst, or cystic degeneration of fibrous dysplasia, may clinically mimic a sarcomatous change. CT and magnetic resonance imaging (MRI) are useful in differentiating malignant lesions from the aforementioned benign cystic bone lesions. Biopsy is performed for histological diagnosis and grading.
The role of radiation in malignant transformation of fibrous dysplasia remains controversial. In a study of 28 cases of fibrous dysplasia with malignant transformation, 13 patients (46%) had received radiation therapy prior to the diagnosis of the malignant change. Generally, radiation is considered not effective in the management of fibrous dysplasia. Where is it possibile that radiation may actually induce sarcomatous change in fibrous dysplasia, it is not recommended in the management of fibrous dysplasia.19,20 Malignancies arising from pre-existing fibrous dysplasia are typically aggressive. Urgent evaluation is needed if rapid swelling and pain occur in a pre-existing fibrous dysplasia lesion.21,22 In treating malignant lesions of the jaws, Russ and Jesse state that surgical extirpation usually implies mandibulectomy or maxillectomy, depending on tumor location and extent.23 The role of adjuvant radiation and chemotherapy remains debatable.19,23 Despite aggressive treatment, local recurrence and distant metastasis are common.16
Diagnosis/patient presentation
All patients should undergo radiological imaging. Plain radiologic features of fibrous dysplasia are nonspecific and vary widely. The typical appearance is that of radiolucent lesions with a homogeneous ground-glass appearance and ill-defined margins. Occasionally the radiograph may reveal predominantly sclerotic lesions with or without accompanying lytic lesions. These nonspecific features make it difficult to differentiate fibrous dysplasia form other conditions such as ossifying osteoma and Paget’s disease.24,25
At our center, CT scan of the craniofacial skeleton using 1-mm sections and three-dimensional reconstruction is routinely performed and is the investigation of choice. From the clinical findings and CT scan, the surgeon is able to obtain useful information regarding tumor size, location, any local invasion or compression. The CT appearance of fibrous dysplasia can be divided into three separate subtypes based on the proportion of ossified tissue and fibrous tissue within the lesion.26 Pagetoid lesions have a mixture of radiodense and radiolucent areas of fibrosis. Sclerotic lesions are homogeneously dense in appearance. Cystic lesions contain one or more lucencies that are surrounded by a dense periphery.
MRI has been suggested some authors as a diagnostic tool for fibrous dysplasia.27 Lesions have been described as areas with a decreased signal as well as sharply demarcated borders on both T1- and T2-weighted images.28 However some authors have concerns regarding the potential of misdiagnosis with MRI.29 The MRI features of fibrous dysplasia do not share the characteristics seen on plain radiography and CT. In fact, the MRI images of fibrous dysplasia often resemble that of tumors. This is particularly so when the lesions show intermediate signal intensities on T1-weighted images and high signal intensities on T2 images, and enhance brilliantly after injection of contrast material. The likelihood of correctly diagnosed fibrous dysplasia by MRI is high only when the signal intensities on both T1- and T2-weighted images are low despite the injection of contrast material.30
Bone scintigraphy may have some role in the diagnosis of fibrous dysplasia. The appearance of fibrous dysplasia on a bone scan is the result of increased tracer uptake in the diseased bone. Radionuclide scan has high sensitivity but low specificity, but it may be used for assessment of the extent of skeletal involvement in polyostotic disease.31 Single-photon emission computed tomography (SPECT) has been reported to be sensitive in the detection of lesions that are not seen on CT.32
Histological samples can be obtained by an isolated incisional biopsy. Alternatively, tissue sample can be obtained at the time of surgical resection of the lesion itself. The caveat of the latter is that decision for a more radical resection cannot hinge on the result of the tissue biopsy. To avoid this scenario, it is recommended to do the biopsy as a separate procedure and wait for the result before the resection. In the scenario where the histologic sample is to be obtained during the operation, and if there are any doubts intraoperatively regarding the nature of the lesion, the resection part of the operation is deferred until final histological results are available. Although the histology of fibrous dysplasia is well established, cytological descriptions are rare. One group reported on fine-needle aspiration cytomorphology of fibrous dysplasia. The smears contained blood, occasional osteoclastic multinucleated giant cells, and frequent C-shaped fibrillary structures with dark central areas and lighter peripheries representing woven bone.33 At present, the role of fine-needle aspiration cytology in fibrous dysplasia is still uncertain.
Biochemical markers such as serum alkaline phosphatase and urinary hydroxyproline are used occasionally to monitor disease progress and the response to the nonsurgical treatment.24
Patient selection
Fibrous dysplasia of the anterior cranial base may compress the optic nerve and cause visual disturbance or even blindness. However controversy surrounds the management of fibrous dysplasia involving encasement of the optic canal, particularly in patients whose vision is normal. Lee et al. suggested that, while radiologic imaging showed that encasement of the optic canal causes narrowing of the canal, this alone does not result in visual loss.34 Thus prophylactic decompression of the optic nerve is not indicated on the basis of the presence of encasement of the optic canal alone without visual symptoms, since the results of such imaging do not correlate with loss of vision. Moreover optic nerve decompression may associate with risks of failure of visual improvement after decompression or even blindness, even though the risks appear to be low in experienced hands.35 In patients with optic canal encasement but without visual symptoms, regular observation with clinical examination and diagnostic imaging is more appropriate. Prophylactic decompression is not advised to be performed as a primary procedure, but as a procedure secondary to resection of the fibrous dysplasia lesion in the anterior skull base during the same operation. On the other hand, therapeutic decompression is advocated in patients with progressive deterioration of vision. For cases with sudden visual loss, it is recommended that decompression should be performed within 1 week of the onset of the symptom to maximize the chance of visual improvement.35
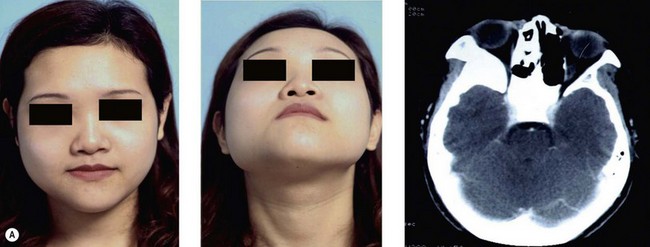
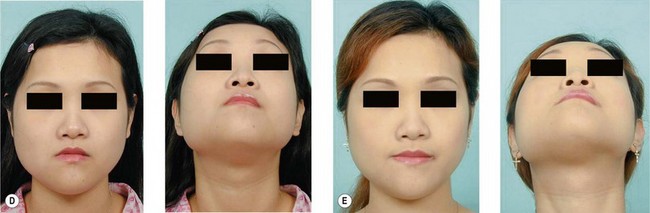
Some patients may present with a more acute clinical scenario. A previously diagnosed fibrous dysplasia may undergo cystic degeneration within the lesion. This usually manifests as pain and rapid enlargement of the lesion (Fig. 15.1). Depending on its location, this may lead to disastrous complications, such as acute optic nerve compression with sudden deterioration of vision. Due to its rapidly progressing nature, cystic degeneration may clinically resemble malignant degeneration. While CT scan or MRI may aid in differentiating the two, sometimes revealing fluid level in the nonmalignant lesion, tissue biopsy may be needed to secure a more definite diagnosis. In cystic degeneration, total excision of the lesion is recommended.24