5 Treatment of Rare Skin Malignancies The skin is the largest organ in the body, and it is not surprising that skin cancer is the most common type of cancer. The skin has a multitude of functions; these include being a protective barrier, a structure of mechanical support, an organ for temperature regulation, a system for excreting toxic substances, an organ for immunologic function, and a sensory organ. While skin cancer usually presents as basal cell and squamous cell cancers or melanoma, cancerous changes can develop in any of the cells that perform the varied functions of the skin. Like the more common skin cancers, most are more common in the Caucasian population, and ultraviolet (UV) exposure is thought to be a risk factor in many of these malignancies. Although these cancers are rare compared to common cutaneous cancers, knowledge of them is important for their treatment. Friedrich Merkel first described Merkel cells in 1875. These slowly adapting mechanoreceptors are derived from the neural crest and reside in the basal layer of the epidermis. Cyril Toker reported a tumor of these cells in 1972, initially describing it as a trabecular skin carcinoma. Six years later, Tang and Toker discovered dense core granules in some of those tumors, which in the skin are only found in Merkel cells.1 The name Merkel cell carcinoma was introduced in 1980. Merkel cell carcinoma is a rare tumor with an incidence of less than 1/100,000 in the United States. Caucasian men are affected most commonly, at an average age of around 70 years. Caucasian patients account for 94% of cases, with an incidence rate 11.3 times greater than African American patients. Over 75% of cases occur in patients over the age of 64 years, with almost half of the cases affecting patients over the age of 74 years. Men are affected about twice as often as women and are diagnosed at an earlier age. Sun exposure (particularly UVB light) seems to play a role, with the highest incidence in the United States occurring in Hawaii, the state with the highest UVB index. The location of tumors correlates with sun-exposed regions, with the head and neck (most commonly the face) affected most often.2 Mucosal lesions account for 4.5% of cases. Arsenic and infrared have also been theorized to have a role in the development of Merkel cell carcinoma.3 Immunocompromised patients are at greater risk, including those with chronic lymphocytic leukemia, acquired immunodeficiency syndrome, and organ transplant patients. Immunocompromised patients develop Merkel cell carcinoma earlier, with a mean age of 53 in transplant patients, and may have worse outcomes.4 The AEIOU mnemonic may be useful for remembering patient demographics. It stands for Asymptomatic, Expanding, Immunosuppression, Older than 50 years, and Ultraviolet exposure in fair-skinned patients.5 Merkel cell carcinoma is difficult to diagnose clinically, lacking distinctive characteristics. The head and neck are most commonly affected, followed by the extremities, and, less commonly, the trunk. It may occur anywhere on the skin or mucosa, and usually presents as a skin toned, red, or purple intradermal nodule that has rapidly grown ( As with most suspicious skin lesions, a biopsy should be performed using punch, incisional, or close-margin excisional technique depending on the lesion location and size. The most common initial clinical suspicion is usually an epidermal cyst,7 but the differential diagnosis includes basal cell carcinoma, squamous cell carcinoma, amelanotic melanoma, adnexal tumors, and cutaneous metastases.3 A dermatopathologist should be requested for analysis of the biopsy. Hematoxylin and eosin (H&E) stains show small, round, blue cells, common in neuroendocrine tumors ( Fig. 5.1 (a) A 73-year-old female with 3 months’ history of nodule on her nasal sidewall that was confirmed to be Merkel cell carcinoma on biopsy. (b) Following excision by Mohs micrographic surgery. The lesion was larger than its clinical appearance. About 5% of Merkel cell carcinomas present in a lymph node with an unknown primary. Half of these are found in cervical nodes and 40% in inguinal nodes, while known primary tumors have positive nodes in the axillary or inguinal region more commonly than in the cervical region.10 Diagnosis in these cases is made by pathology with the same stains mentioned earlier. At the time of diagnosis, two-thirds of patients have local disease, one-quarter have lymph node involvement, and 7% have distant metastases.11 A thorough review of systems should be obtained, and a complete physical examination including skin evaluation and palpation for lymphadenopathy should be performed. Sixteen to 24% of patients have palpable regional nodes.7,12 The National Comprehensive Cancer Network (NCCN) recommends imaging studies for patients with clinically positive nodes. Positron emission tomography–computed tomography (PET-CT) is preferred, which is sensitive and specific, but CT or magnetic resonance imaging (MRI) is an option if PET-CT is not available.9 PET-CT leads to a change in management in one-third of patients. Upstaging is relatively common (22% of total patients), most often with a finding of bone metastases, but downstaging also occurs in 5% of the cases. The PET stage is associated with survival, making it a useful test for prognosis.13 Another indication for imaging is when small cell carcinoma of the lung with skin metastasis is a possible diagnosis. For example, when a biopsy is suspicious for Merkel cell carcinoma on histology, but is CK-20 negative, the NCCN recommends CT, MRI, or PET-CT to evaluate for a different primary cancer.9 In 2008, Feng et al identified a polyomavirus present in 80% of Merkel cell tumors, compared to about 10% of control skin samples. This virus was named Merkel cell polyomavirus (MCV).14 This virus is a part of normal human flora, and infected patients are asymptomatic. Most people acquire MCV by middle age. The virus is then integrated into the genome.15 MCV infection and high levels of antibodies against MCV are associated with a higher risk of developing Merkel cell carcinoma; however, these factors are associated with better survival rates.16 Fig. 5.2 (a) Hematoxylin and eosin (H&E) stain of Merkel cell carcinoma shows small round blue cells. (b) High-power view. Fig. 5.3 Positive cytokeratin 20 stain, found in nearly all cases of Merkel cell carcinoma. The tumor, node, metastasis (TNM) Staging System is used for Merkel cell carcinoma, as described by the American Joint Committee on Cancer. T1 indicates tumors less than or equal to 2 cm, T2 tumors 2 to 5 cm, and T3 tumors over 5 cm. N0 indicates no nodal involvement, and N1 indicates metastases to regional nodes. M0 means no metastases, and M1 means distant metastasis (beyond lymph nodes).17 Stage 0 tumors are in situ. Stages I and II are local, with stage II including tumors greater than or equal to 2 cm. Stage III indicates nodal involvement, and stage IV tumors have distant metastasis. Distant metastases occur most commonly in the liver, followed by the lungs, bones, and brain.3 The NCCN guidelines recommend wide excision with 1- to 2-cm margins, removing the specimen down to fascia. Some studies, however, found no change in local recurrence with 2-cm margins compared to 1 cm, and therefore some authors advocate for 2-cm margins only in cases where the primary tumor is greater than 2 cm.3,12 Mohs or modified Mohs (Mohs excision, followed by a 0.5- or 1-cm margin after the final section) is also acceptable in areas of cosmetic concern, such as the face. The central portion of the specimen should be sent as a permanent specimen. The wound should be closed with the goal of rapid healing if radiation therapy is planned9 ( Sentinel lymph node biopsy (SLNB) should be performed in all patients at the time of the wide excision, both for staging and for therapeutic reasons ( Fig. 5.4 (a) Merkel cell carcinoma excision after biopsy. The scar was marked with 2-cm margins and injected with methylene blue for sentinel lymph node biopsy. (b) After wide excision down to investing fascia, the wound was covered. (c) Sentinel lymph nodes were identified in the axilla. These were identified by lymphoscintigraphy using a gamma probe and methylene blue. For patients with a positive sentinel lymph node or positive clinical nodes on exam, therapeutic regional lymph node dissection is recommended. Improved survival is seen in some studies.19 Type I interferons have been found to inhibit the proliferation of MCV positive Merkel cell carcinoma, inducing apoptosis in vitro and in vivo.20 More research is needed before treatment recommendations can be made. Radiation to the primary site and/or the regional nodes may decrease local recurrence and improve disease-free survival.6,21,22 This is controversial since other studies show no change in recurrence or overall survival.12,23 The NCCN recommends radiation to the primary site (and the nodal basin in head and neck tumors) for patients with negative SLNB. If SLNB is not obtained or is positive, the primary site and nodal basin should be radiated.9 Radiation should be given as soon as the surgical wounds have healed. Traditional chemotherapy regimens including carboplatin and etoposide are poorly tolerated and have not been shown to decrease mortality; they have therefore not been recommended for routine use by the NCCN9. Immunotherapy has recently shown promising results in both MCV positive and MCV negative patients. These new medications include pembrolizumab and nivolumab, which inhibit programmed death-1 (PD-1), and avelumab, which inhibits programmed death ligand-1 (PD-L1). Immunotherapy should be initiated early in patients with metastatic disease.24 The overall 5-year survival rate is poor, at only about 50%. Immunosuppressed patients have an even worse prognosis, with only 43% alive 3 years after their diagnosis.7 Worse survival rates are seen in men and the elderly.19 Tumors on the limb are often diagnosed at an earlier stage than those on the trunk, and patients with tumors of the upper extremity have improved survival.25 Most patients present with stage I or stage II disease. Staging is indeed predictive of survival, with patients diagnosed with stage I disease having an 86% 2-year survival rate, compared to 32% in patients with stage II disease.2 Although patients with an unknown primary are stage III due to nodal involvement, these patients have a better survival rate than patients with a known primary who are stage III.10 Node status and the corresponding pathologic stage are the most important predictors for prognosis. SLNB itself may be therapeutic in addition to being prognostic, with SLNB patients having improved overall survival rate compared to patients with positive nodes and also those who did not have a lymph node evaluation. This is particularly true for tumors that were not on the head and neck,22,26 whereas head and neck tumors have less predictable drainage patterns, possibly leading to a high false-negative SLNB rate.18 Up to half of patients have local or regional recurrence, usually in the nodal basin.7,12 This occurs in an average of 9 months, with over 90% of recurrences occurring within 2 years. In patients with distant metastases, average survival is less than 1 year.12 Patients should be evaluated every 3 to 6 months for the first 2 years after treatment, and then at least yearly.9 Merkel cell carcinoma is a rare, but locally and regionally aggressive skin cancer that most commonly affects elderly white males. It is difficult to diagnose clinically, and histological diagnosis is assisted by special stains. Merkel cell carcinoma should be treated with wide local excision using at least 1-cm margins. SLNB should be mandatory, given it determines staging, which predicts prognosis. Radiation therapy is often indicated, while chemotherapy has not proven to be effective. MCV is often present in the tumor, but has yet to be effectively targeted therapeutically. Dermatofibrosarcoma protuberans (DFSP) is a malignant cutaneous tumor characterized by a low risk of distant metastasis but a high local recurrence rate if not treated appropriately. It is a mesenchymal tumor, stimulated by mutations in stem cells in skin appendages.27 DFSP was first described by Taylor in 1890 as a sarcoma “resembling in some aspects keloids.” It was later given its name by Hoffman in 1925.28 DFSP has an incidence of 1 to 4 cases per million people per year in the United States, accounting for 0.1% of all cancers.29 It affects males and females nearly equally, with a slight predominance in women. Among American Caucasians, the highest incidence is in Hawaii29; however, a quarter of cases occur in non-Caucasian patients, with African Americans being disproportionately affected. DFSP is most commonly diagnosed in patients 30 to 50 years old,29 but up to 20% of cases occur in pediatric patients. Congenital lesions are also reported.30 DFSP lacks defining features, and is often present for many years before it is biopsied and diagnosed. It usually presents as an asymptomatic, firm plaque or nodule that has been slowly enlarging for a long time, sometimes with a recent rapid growth period.29 Some patients describe a history of trauma preceding the lesion, but this has not been correlated with development of DFSP.30 DFSP may be skin-toned, red-blue, brown-yellow, erythematous, sclerodermiform, telangiectatic, or atrophic. It is adherent to the skin but usually freely mobile over deeper structures.31 Long-standing lesions may become painful or develop ulcerations.28 DFSP can present anywhere, but most commonly is found on the trunk. Thirty percent of tumors appear in the extremities (upper more often than lower), and 15% in the head and neck. The distribution in children is similar.32 Incisional or excisional biopsy should be performed. H&E stain shows a uniform proliferation of spindle cells with low mitotic activity. These form a characteristic “storiform” or “cartwheel” pattern ( Fig. 5.6 Storiform pattern of dermatofibrosarcoma protuberans. The differential diagnosis may also include neurofibroma, leiomyoma, epidermal cyst, malignant melanoma, basal cell carcinoma, keloid, desmoids, Kaposi’s sarcoma, and sarcoidosis.28 In children, DFSP may be confused with vascular malformations and hamartomas, as well as congenital aplasia cutis, dermoid cyst or teratoma, and infantile fibromatosis.32 A thorough history and physical exam is usually sufficient prior to treatment of DFSP, given it is generally a local disease. DFSP metastasizes in only 1% of patients, mostly to the lungs. It does not commonly spread to the lymph nodes, and therefore SLNB is not recommended. MRI accurately depicts the depth of lesions, but underestimates the lateral extent.33 The NCCN does not recommend any diagnostic imaging.34 Several variants exist, including the Bednar’s tumor, a pigmented variant making up 1% of DFSP cases, as well as myxoid, myoid, granular cell, neuorfibromatous, sclerotic, giant cell fibroblastoma, and atrophic. A more aggressive subtype, fibrosarcomatous DFSP (DFSP-FS), has more frequent mitoses and areas of cellular atypia, with a herringbone pattern. This variant accounts for 10 to 15% of cases and is aggressive, with higher local recurrence rates, more frequent metastasis, and greater mortality.35 Over 90% of cases have supernumerary ring chromosomes or translocations (t[17;22] [q22;q13]), combining parts of chromosomes 17 and 22. This results in an oncogenic fusion of collagen type I alpha 1 gene (COL1A1) with platelet-derived growth factor B-chain gene (PDGFB), resulting in lack of regulation of production of the PDGFB protein.36 In turn, the PDGFB receptor, a tyrosine kinase, is overstimulated. The chromosomal changes are confirmed with karyotype analysis and fluorescence in situ hybridization (FISH), and the gene fusion is discovered with polymerase chain reaction (PCR).27 Fig. 5.7 Positive CD34 stain, typically found in dermatofibrosarcoma protuberans. Several staging systems have been devised for DFSP, but none accurately predict outcomes.27 The American Musculoskeletal Tumor Society system uses histologic grade, local extension, and metastases. The American Joint Committee on Cancer includes tumor size and depth, lymph node involvement, and distant metastasis. The Short German Guidelines are also available.28 Ninety-nine percent of cases present with local or regional involvement only.29 Treatment consists of complete excision of tumor. This can be difficult due to projections coursing microscopically much further than are visible clinically, sometimes over 10 cm.37 Recurrence rates after wide local excision are reported to be as high as 60%.28 With careful analysis of margins, recurrence rates after wide local excision can be as low as 1%, but may require multiple excisions and delayed wound closure.38 The NCCN recommends margins of 2 to 4 cm when performing wide local excision, taking specimens down to investing fascia.34 Due to the difficulty in obtaining clear margins, Mohs micro-graphic surgery (MMS) has become the standard of care for DFSP, particularly in the head and neck. Recurrence rates after Mohs controlled excision are reported at about 0 to 1%.39,40 Mohs defects are on average four times larger than the clinical lesion.40 Treating all patients with conventional wide excision results in excessive resection in 80% of patients and inadequate resection in 5%, supporting MMS as the treatment of choice.31 While MMS is effective, it takes longer, requires a dermatologic surgeon who is familiar with DFSP frozen sections, and is expensive. Frozen sections can be difficult to interpret for an inexperienced dermatopathologist, with about 90% accuracy when compared to permanent pathology.38 Many surgeons send the final stage as a permanent specimen for confirmation of negative margins (modified Mohs), especially in recurrent cases. Reconstruction with tissue rearrangement should be delayed until negative margins are confirmed34 ( DFSP is radiosensitive; however, due to the reasonably low recurrence rates with adequate resection, radiation therapy should be reserved for unresectable, recurrent, or metastatic cases.34 Patients with microscopically positive margins have a 33% recurrence rate.41 Treating all of these patients can potentially confer more risk than benefit, as the majority do not go on to develop local or distant metastases. Patients with the COL1A1-PDGFB fusion gene may benefit from imatinib mesylate, a tyrosine kinase inhibitor. Up to two-thirds of patients will have tumor regression. Neoadjuvant therapy may make reconstruction simpler, but can change the appearance on histology by decreasing cellularity.27 While this drug is well tolerated by most, patients should be screened for the fusion gene prior to treatment to prevent side effects in patients who will not have a response.42 The NCCN recommends imatinib in unresectable, recurrent, or metastatic cases.34 Traditional chemotherapy is only recommended in patients with metastatic disease.34 Although DFSP is known for high recurrence rates, distant spread is rare, with only about 1% of patients developing distant metastasis. Metastases usually follow local recurrences, often in DFSP-FS.35 Patients have an overall 5-year survival of 99%, and a disease-specific 5-year survival of over 99%.29 Recurrent disease is associated with decreased disease-free survival.41 Patients with DFSP are at increased risk for a second DFSP lesion, as well as for subsequent primary malignancies of other types. These are most often skin cancers, but also include breast cancer.43 Patients should be seen for follow-up every 6 to 12 months. Twenty percent of recurrences occur over 3 years after resection of the primary lesion.27 Because of the possibility for late recurrence, as well as the increased risk of other cancers, follow-up should continue indefinitely. DFSP is a locally aggressive skin cancer that affects men and women in their 30s to 50s, with an increased prevalence in African Americans. It often presents in a delayed fashion due to its slow growth and indistinct form, and is diagnosed with a biopsy using H&E and immunohistochemistry. Treatment consists of complete excision, either by wide local excision or with MMS. Imatinib may be useful in some patients with a specific gene fusion. Overall survival is excellent, and distant metastasis is rare. Cutaneous angiosarcoma is a rare mesenchymal malignancy of vascular origin. It is one of the most aggressive skin cancers, with high rates of local recurrence and distant metastases, as well as high mortality rate. Cutaneous sarcomas account for 5% of skin malignancies, and angiosarcomas account for less than 2% of all soft tissue sarcomas.44,45 More than half of angiosarcomas occur in the skin and superficial subcutaneous tissues,46 with an overall incidence of 0.01/100,000 per year.47 Cutaneous angiosarcoma occurs in three settings: spontaneously, following radiation therapy, and in the setting of chronic lymphedema (known as Stewart–Treves syndrome). The spontaneous category includes malignant degeneration of a vascular malformation.48 Other possible contributing factors are excessive UV light,49 immunosuppression,45 and polyvinyl chloride, arsenic, or thorium dioxide exposure.50 Spontaneous cases affect men more often; however, many of the cases related to radiation and lymphedema occur in women after breast cancer treatment. Cutaneous angiosarcoma usually occurs in Caucasian patients over 50 years old, peaking in the seventh or eighth decade of life. It has also been reported in children as young as 6 weeks, accounting for 0.3% of pediatric sarcomas.51 Cutaneous angiosarcoma often looks like a bruise initially, which seems to enlarge over time. It may become nodular, painful, edematous, friable, or ulcerate.46 Patients often have symptoms for several months prior to diagnosis.46 At the time of diagnosis, the size of the lesion can range from less than 1 cm to over 10 cm.45,48 The differential diagnosis includes pyogenic granuloma, Kaposi’s sarcoma, lymphoma, malignant melanoma, recurrent breast cancer, or radiation-related morphea.48,52 Lesions occur most commonly on the scalp and face, followed by the trunk, lower extremity, and upper extremity.45 The high propensity for the scalp is thought to be influenced by increased UV exposure to this area combined with a relatively increased vascularity.48 Lesions on the breast are most commonly associated with prior radiation for treatment of breast cancer, and lesions on the arm are often in the setting of Stewart–Treves syndrome. Diagnosis is made by excisional or incisional biopsy. Histology shows vascular channels lined by endothelial cells, often dissecting collagen in the dermis and extending into the subcutaneous tissue46,52 ( A careful history and physical exam should be performed, searching for multifocal or satellite disease, which is present in over one-third of patients, as well as locoregional and distant metastases. Metastasis eventually occurs in over 50% of patients, with metastasis most commonly to the lungs, as well as bone, liver, spleen, and less commonly to lymph nodes.48 MRI may be used as an adjunct, but it shows greater local involvement than seen grossly.46 MRI, CT, or PET-CT can detect local or distant metastases.
5.1 Introduction
5.2 Merkel Cell Carcinoma
5.2.1 Background
5.2.2 Epidemiology
5.2.3 Diagnosis
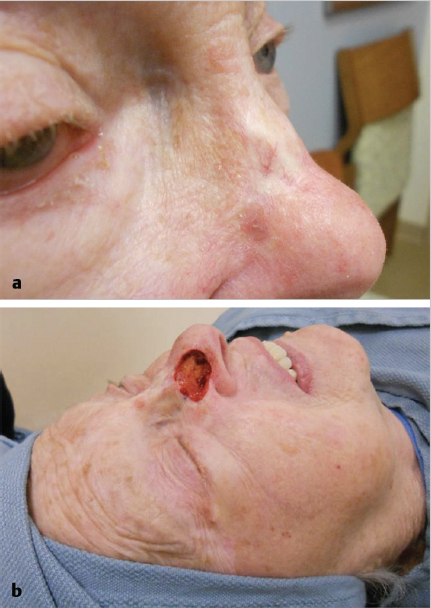
![]() Fig. 5.1a, b). The epidermis may be ulcerated. Most tumors are less than 2 cm at the time of diagnosis.6
Fig. 5.1a, b). The epidermis may be ulcerated. Most tumors are less than 2 cm at the time of diagnosis.6
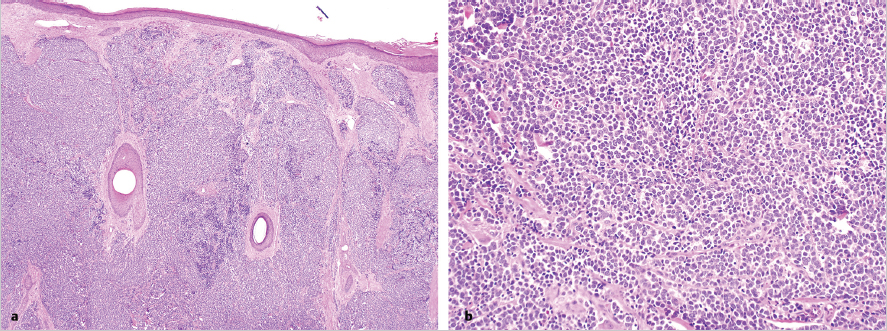
![]() Fig. 5.2a, b). Immunohistochemistry must be completed to confirm the diagnosis, since metastatic small cell lung carcinoma (SCLC) can be indistinguishable on H&E. Cytokeratin-20 (CK-20) is found in 89 to 100% in cases of Merkel cell carcinoma, and is uncommonly seen in SCLC (
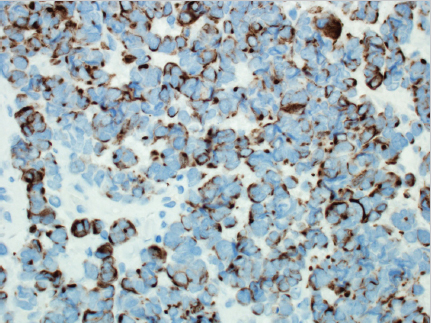
Fig. 5.2a, b). Immunohistochemistry must be completed to confirm the diagnosis, since metastatic small cell lung carcinoma (SCLC) can be indistinguishable on H&E. Cytokeratin-20 (CK-20) is found in 89 to 100% in cases of Merkel cell carcinoma, and is uncommonly seen in SCLC (![]() Fig. 5.3). Merkel cell carcinoma stains are positive for neuro-filaments in most cases, but SCLC stains are not. Conversely, thyroid transcription factor 1 is positive in most SCLCs, but it is seen rarely in Merkel cell carcinoma.8 For lesions that test equivocally, additional stains including chromogranin, synaptophysin, CD56, and neuron-specific enolase may be used to evaluate for other diagnoses.9
Fig. 5.3). Merkel cell carcinoma stains are positive for neuro-filaments in most cases, but SCLC stains are not. Conversely, thyroid transcription factor 1 is positive in most SCLCs, but it is seen rarely in Merkel cell carcinoma.8 For lesions that test equivocally, additional stains including chromogranin, synaptophysin, CD56, and neuron-specific enolase may be used to evaluate for other diagnoses.9
5.2.4 Workup
5.2.5 Merkel Cell Polyomavirus
5.2.6 Staging
5.2.7 Surgical Treatment
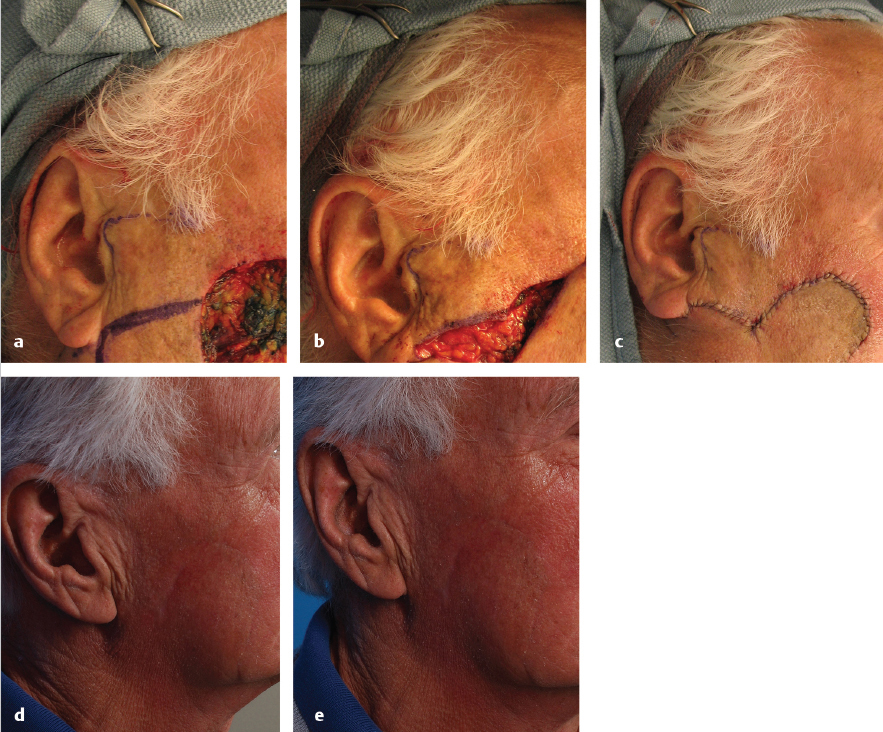
![]() Fig. 5.4a, b).
Fig. 5.4a, b).
![]() Fig. 5.4c). Sentinel node status is the most important information for staging (
Fig. 5.4c). Sentinel node status is the most important information for staging (![]() Fig. 5.5). Merkel cell carcinoma spread is generally lymphatic rather than hematogenous. Lymph nodes are commonly involved at the time of diagnosis, even in relatively small and/or superficial tumors with up to 37% having a positive sentinel node.6,18 In patients without palpable lymphadenopathy, micrometastases are found in 23%.12 Positive sentinel nodes are more common in tumors that spread below the dermis, and in older patients.18 Sentinel nodes should be evaluated using the appropriate stains as mentioned earlier. Pathology should also note the amount of tumor burden and whether the tumor spreads beyond the capsule of the lymph node.9
Fig. 5.5). Merkel cell carcinoma spread is generally lymphatic rather than hematogenous. Lymph nodes are commonly involved at the time of diagnosis, even in relatively small and/or superficial tumors with up to 37% having a positive sentinel node.6,18 In patients without palpable lymphadenopathy, micrometastases are found in 23%.12 Positive sentinel nodes are more common in tumors that spread below the dermis, and in older patients.18 Sentinel nodes should be evaluated using the appropriate stains as mentioned earlier. Pathology should also note the amount of tumor burden and whether the tumor spreads beyond the capsule of the lymph node.9
5.2.8 Radiation Therapy
5.2.9 Chemotherapy
5.2.10 Prognosis
5.2.11 Summary
5.3 Dermatofibrosarcoma Protuberans
5.3.1 Background
5.3.2 Epidemiology
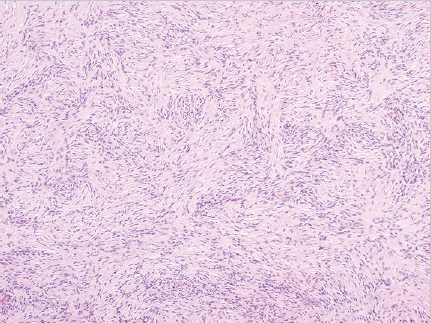
5.3.3 Diagnosis
![]() Fig. 5.6), with projections invading underlying fat, fascia, muscle, and even bone, like tentacles. This creates a honeycomb appearance within subcutaneous fat. It may be difficult to distinguish DFSP from dermatofibroma, fibrosarcoma, atypical fibroxanthoma (AFX), and nodular fasciitis.28 Immunohistochemistry can be helpful. CD34 is often strongly expressed in DFSP (
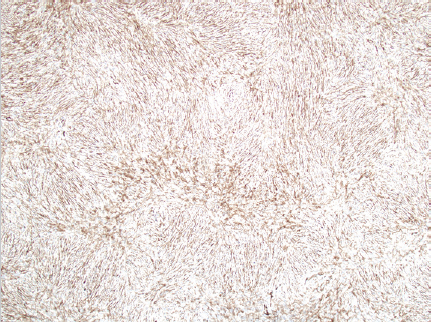
Fig. 5.6), with projections invading underlying fat, fascia, muscle, and even bone, like tentacles. This creates a honeycomb appearance within subcutaneous fat. It may be difficult to distinguish DFSP from dermatofibroma, fibrosarcoma, atypical fibroxanthoma (AFX), and nodular fasciitis.28 Immunohistochemistry can be helpful. CD34 is often strongly expressed in DFSP (![]() Fig. 5.7) and is used to differentiate it from dermatofibromas, which do not express CD34. Conversely, dermatofibromas express coagulation factor XIIIa, while DFSP does not.28 Other stains including stromolysin-3, S100, CD68, and apolipoprotein D may also be used to rule out other diagnoses.
Fig. 5.7) and is used to differentiate it from dermatofibromas, which do not express CD34. Conversely, dermatofibromas express coagulation factor XIIIa, while DFSP does not.28 Other stains including stromolysin-3, S100, CD68, and apolipoprotein D may also be used to rule out other diagnoses.
5.3.4 Workup
5.3.5 Subtypes
5.3.6 Chromosomal Abnormalities
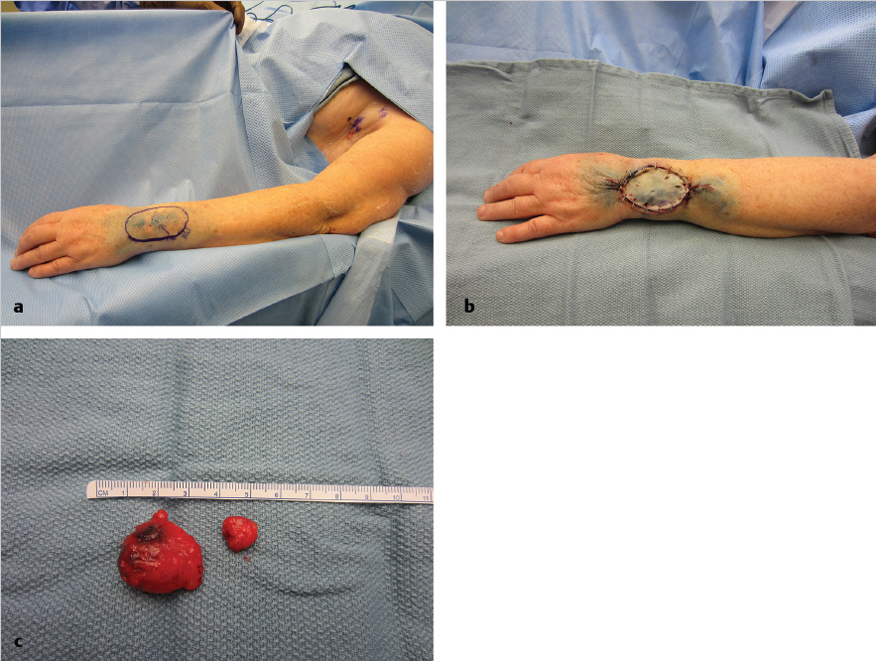
5.3.7 Staging
5.3.8 Surgical Treatment
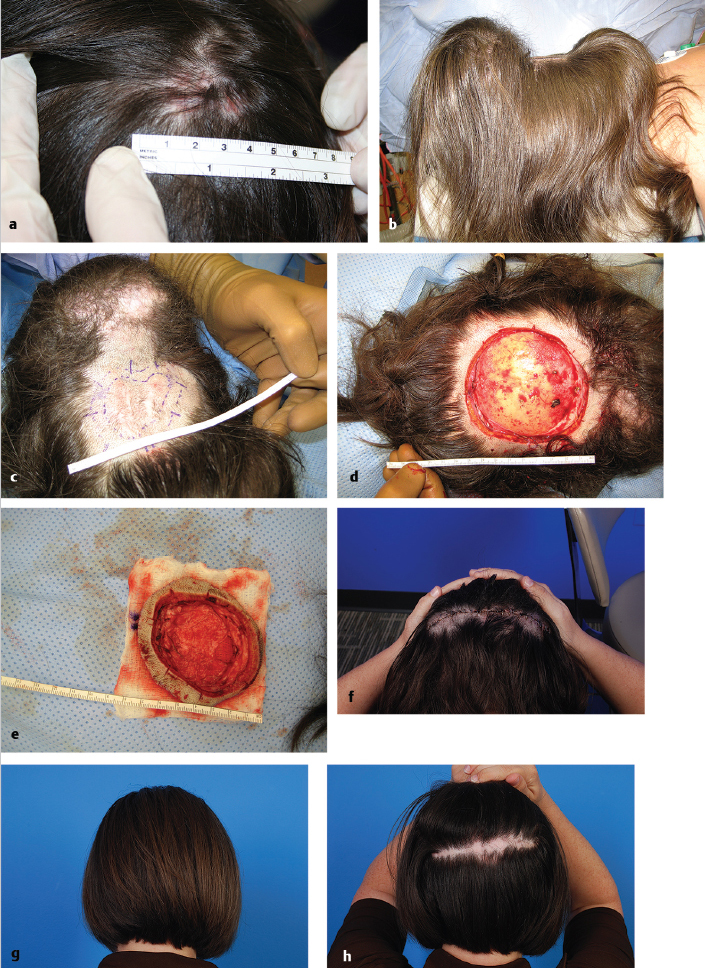
![]() Fig. 5.8a-h).
Fig. 5.8a-h).
5.3.9 Radiation Therapy
5.3.10 Chemotherapy
5.3.11 Prognosis
5.3.12 Summary
5.4 Cutaneous Angiosarcoma
5.4.1 Background
5.4.2 Epidemiology
5.4.3 Diagnosis
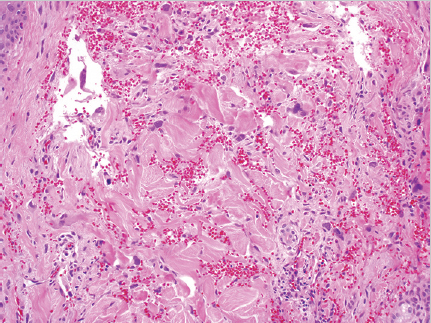
![]() Fig. 5.9). Blood-filled channels or clefts are sometimes present with hemorrhage and necrosis.52 Lesions range from well to poorly differentiated. On histology, the lesion may be difficult to distinguish from other vascular and epithelial tumors including spindle cell carcinoma, malignant melanoma, and AFX; therefore, immunohistochemistry, and in particular vascular markers, should be used.53 Cutaneous angiosarcoma often stains positive for CD31 and Ets family transcription factor (ERG).54 Less sensitive stains include vascular endothelial growth factor receptor 3 (VEGFR-3), factor VIII, and D2–D40 monoclonal antibody.47,54 Angiosarcomas are usually negative for cytokeratin, S100, and desmin. Cutaneous angiosarcoma subtypes including the epithelioid subtype are diagnosed by histology and immunohistochemistry.
Fig. 5.9). Blood-filled channels or clefts are sometimes present with hemorrhage and necrosis.52 Lesions range from well to poorly differentiated. On histology, the lesion may be difficult to distinguish from other vascular and epithelial tumors including spindle cell carcinoma, malignant melanoma, and AFX; therefore, immunohistochemistry, and in particular vascular markers, should be used.53 Cutaneous angiosarcoma often stains positive for CD31 and Ets family transcription factor (ERG).54 Less sensitive stains include vascular endothelial growth factor receptor 3 (VEGFR-3), factor VIII, and D2–D40 monoclonal antibody.47,54 Angiosarcomas are usually negative for cytokeratin, S100, and desmin. Cutaneous angiosarcoma subtypes including the epithelioid subtype are diagnosed by histology and immunohistochemistry.
5.4.4 Workup
Plastic Surgery Key
Fastest Plastic Surgery & Dermatology Insight Engine