The Porphyrias: Introduction
The porphyrias are among the most intriguing human diseases. Widely variable, even bizarre in their clinical manifestations, these disorders of porphyrin or porphyrin-precursor metabolism result from aberrations in the control of the heme biosynthetic pathway. Heme is essential for oxygen binding and transport (as in hemoglobin and myoglobin), for electron transport (as in cytochromes), and for monooxygenases such as cytochrome P450. Chlorophyll, a magnesium-chelated porphyrin, is another important tetrapyrrole that is critical for photosynthesis, the specialized energy-storing system found in plants in which the conversion of light energy into stabilized chemical energy is achieved with a sequence of oxidation-reduction reactions. The corrin ring, a cobalt-chelated tetrapyrrole, is a major constituent of vitamin B12, the lack of which results in pernicious anemia. Therefore, porphyrins are ubiquitous and essential biochemical constituents of living beings. The biologic importance of the porphyrins and their iron complexes lies in their capacity to facilitate metabolic reactions, either as oxidative components in the metabolism of steroids, drugs, and environmental chemicals or by enhancing gas exchange, such as oxygen and carbon dioxide, between the environment and the tissues of the body.
Daily synthesis of porphyrins and heme in humans occurs in amounts sufficient to provide for the body’s metabolic requirements. The control of heme synthesis is so precise that, under normal circumstances, only microgram quantities or less of pathway intermediates are present in plasma, red blood cells (RBC), urine, and stool (Table 132-1).
Porphyrins or Precursors | Urine (μg/24 h) | Red Blood Cells (μg/100 mL Packed Cells) | Plasma (μg/100 mL) | Feces (μg/g Dry Weight) |
|---|---|---|---|---|
δ-Aminolevulinic acid (ALA) | <4,000 | — | 15–23 | — |
Porphobilinogen (PBG) | <1,500 | — | — | — |
Uroporphyrin (URO) | <40 | 0–2.0 | 0–2 | 10–50 |
Coproporphyrin (COPRO) | <280 | 0–2.0 | 0–1 | 10–50 |
Protoporphyrin (PROTO) | Absent | <90 | 0–2 | 0–20 |
X-porphyrin | Absent | Absent | Absent | Trace |
Isocoproporphyrin (ISOCOPRO) | Absent | Absent | Absent | — |
(nmolL/Day) | (nmol/dL) | (nmol/dL) | (nmol/g Dry Weight) | |
ALA | 8.8 × 103 | — | 1.1–1.8 × 102 | — |
PBG | 4.6 × 104 | — | — | — |
URO | <40 | — | 24 | — |
COPRO | <280 | — | 46 | — |
PROTO | 0 | 285 | 0.5 | 134 |
The porphyrias are a clinically and genetically heterogeneous group of metabolic diseases, which result from an either inherited or acquired dysfunction of enzymes crucial for heme biosynthesis (Fig. 132-1). Heme synthesis is controlled by eight enzymes along the heme biosynthetic pathway. Deficient activity of seven of these eight enzymes can give rise to a specific type of porphyria (Fig. 132-1).1–3 A gain of function of the first enzyme in the pathway, δ-aminolevulinic acid synthase (ALAS) is responsible for X-linked dominant protoporphyria (XLDPP), a recently recognized type of porphyria.4 Each of these enzymes will be discussed separately below, in the context of the corresponding type of porphyria.
Figure 132-1
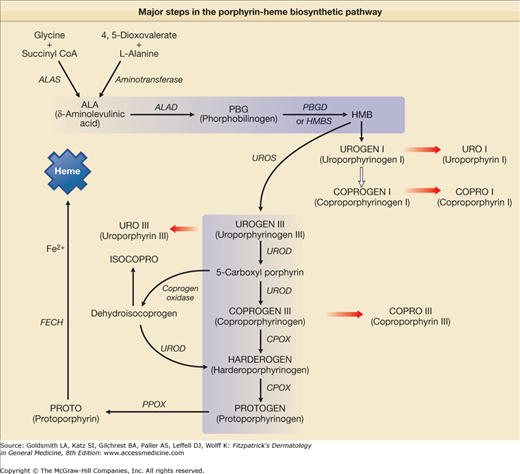
Major steps in the porphyrin-heme biosynthetic pathway. δ-Aminolevulinic acid (ALA) can be formed from glycine and succinyl coenzyme A (SCoA), which is the primary source in mammalian systems and is catalyzed by the mitochondrial enzyme ALA synthase (ALAS). Two molecules of ALA form the monopyrrole porphobilinogen (PBG) in a reaction catalyzed by the enzyme ALA dehydratase (ALAD). Four molecules of PBG are converted by PBG deaminase (PBGD) also known as hydroxymethylbilane synthetase (HMBS) to a linear tetrapyrrole, hydroxymethylbilane (HMB), which can cyclize spontaneously to form uroporphyrinogen (UROGEN) I. The four acetyl groups of UROGEN I are sequentially decarboxylated by UROPORPHYRINOGEN decarboxylase (UROD) to form coproporphyrinogen (COPROGEN) I. HMB can also be converted to UROGEN III by the enzyme UROPORPHYRINOGEN III synthase (UROS). In this reaction, one of the monopyrrole rings is “flipped over,” which alters the sequence of the side chains. The acetyl groups of UROGEN III are sequentially decarboxylated by UROD to form COPROGEN III. COPROGEN III is converted to protoporphyrinogen (PROTOGEN) IX by the enzyme COPROPORPHYRINOGEN oxidase (CPOX), which oxidatively decarboxylates each of the propionyl groups. PROTOGEN IX is converted to protoporphyrin (PROTO) IX by PROTOPORPHYRINOGEN oxidase (PPOX). PROTO IX is converted to heme by ferrochelatase (FECH), which catalyzes the insertion of ferrous iron into the molecule.
Biochemically, the different porphyrias are characterized by particular patterns of accumulation and excretion of porphyrins and/or their precursors. In general, the porphyrins excreted in a specific type of porphyria are the irreversibly oxidized substrate(s) of the deficient enzyme (Table 132-2). These intermediates, when present in excess amounts, exert toxic effects that are responsible for the cutaneous and neurological signs and symptoms of clinically overt porphyria. The porphyrias are of particular dermatologic interest because several types exhibit cutaneous manifestations that may permit diagnosis from clinical signs alone.
Enzyme | Tissue | Enzymatic Defect in Porphyria |
|---|---|---|
| δ-Aminolevulinic acid synthase 1 (ALAS1) | Liver, kidney, fibroblasts, and lymphocytes | Increased in AIP, HCP, VP |
| δ-Aminolevulinic acid synthase 2 (ALAS2) | Erythrocytes | Increased in XLDPP |
| ALA dehydratase (ALAD) | Erythrocytes, liver, and kidney | Decreased in lead intoxication |
| Decreased in ALA dehydratase deficiency porphyria | ||
| Porphobilinogen deaminase (PBGD) | Erythrocytes, liver, fibroblasts, lymphocytes, and amnion cells | Decreased in AIP |
| Uroporphyrinogen III synthase (UROS) | Erythrocytes and fibroblasts | Decreased in CEP |
| Uroporphyrinogen decarboxylase (UROD) | Erythrocytes and liver | Decreased in all tissues in hereditary or familial PCT and HEP |
| Only decreased in the liver in acquired or sporadic PCT | ||
| Coproporphyrinogen oxidase (CPOX) | Fibroblasts, lymphocytes, and liver | Decreased in HCP |
| Protoporphyrinogen oxidase (PPOX) | Liver and fibroblasts | Decreased in VP |
| Ferrochelatase (FECH) | Bone marrow and fibroblasts | Decreased in EPP and lead poisoning |
Porphyrin-Heme Biosynthesis
The major sites of heme synthesis in the body are the bone marrow and the liver. Heme is the prosthetic group for a number of proteins, including among others hemoglobin, myoglobin, mitochondrial cytochromes, microsomal cytochromes (including cytochrome P450), catalase, peroxidase, tryptophanpyrrolase, prostaglandin endoperoxide synthase, and the soluble form of guanylate cyclase. Approximately 85% of heme synthesis occurs in the bone marrow where it is utilized for the production of hemoglobin; the majority of the rest occurs in the liver for making cytochrome P450, catalase, and various mitochondrial cytochromes. Heme is a critical cellular constituent essential for a variety of metabolic processes, primarily because of its unique ability to take up and release oxygen and to facilitate electron transport. Heme synthesis is regulated by the interplay of a number of factors and is directly dependent upon its concentration within cells and upon the requirements of the cell for production of the various hemoproteins described above. Many of these have rapid turnover times (minutes to hours), thereby necessitating continuously high rates of hepatic heme synthesis. For example, cytochrome P450, an important membrane-bound enzyme in the liver involved in the detoxification and metabolism of drugs, has a half-life of 90–180 minutes.
Heme synthesis begins in the mitochondrion of the cell where succinate and glycine (single molecules of glycine and succinyl CoA) are conjugated by ALAS to form a five-carbon aminoketone, δ-aminolevulinic acid (ALA). This reaction requires pyridoxal 5′-phosphate as a cofactor. The regulation of heme biosynthesis (and indeed the ability to synthesize heme at all) is dependent upon the serial interaction of eight intracellular enzymes (Figs. 132-1 and 132-2). The first, as well as the last three enzymes involved, coproporphyrinogen oxidase (CPOX), protoporphyrinogen oxidase (PPOX), and ferrochelatase (FECH) are mitochondrial, whereas the remaining enzymes including δ-aminolevulinic acid dehyratase (ALAD), porphobilinogen deaminase (PBGD), uroporphyrinogen synthase (UROS), and uroporphyrinogen decarboxylase (UROD) are localized in the cytosol. The degradation of heme is catalyzed by the microsomal enzymes NADPH-cytochrome C reductase, heme oxygenase 1 and 2. Heme oxygenase 1 is a -signature of oxidative tissue injury and is a potent antioxidant. Heme oxygenase 2 is an oxygen sensor. The metabolic by-products of these enzymes are the linear tetrapyrrole biliverdin IXα and carbon monoxide and also possess antioxidant properties. cDNAs for each of the eight enzymes involved in heme biosynthesis as well as the enzymes responsible for heme catabolism have been cloned in mammalian cells.1–3 Abnormal regulation of heme synthesis may result from defects in enzymes of the synthetic pathway, and this may occur as the result of inherited and/or environmental factors leading to the accumulation in the body of one or more heme pathway intermediates, such as the porphyrins or their precursors. Although a diagnosis of porphyria can often be made from a careful medical history (including the family history) and physical examination, the definitive diagnosis requires measurement of: (1) porphyrins and/or porphyrin precursors in urine, stool, plasma, or RBCs, or (2) the residual activity of specific enzymes in the heme pathway, or (3) the identification of mutations in the genes encoding these enzymes (Fig. 132-1). Modern molecular biological techniques have helped to clarify the mechanistic basis of the porphyrias.3–8 The steps involved in the biosynthesis of porphyrins and heme and their regulatory control are summarized in Figs. 132-1, 132-2, and 132-4.
Figure 132-2
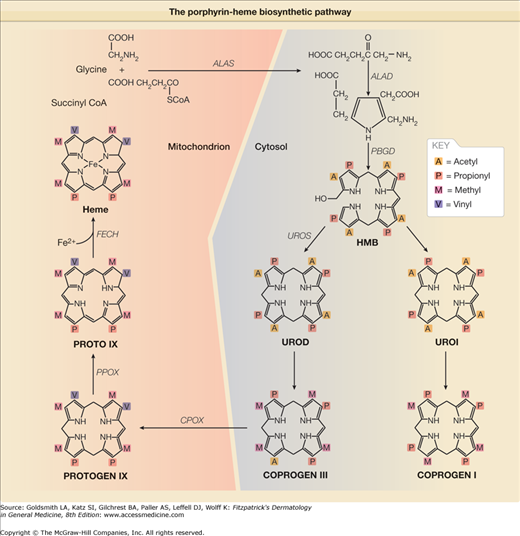
The porphyrin-heme biosynthetic pathway (with structural components). (See also Figure 132-1.)
Figure 132-3
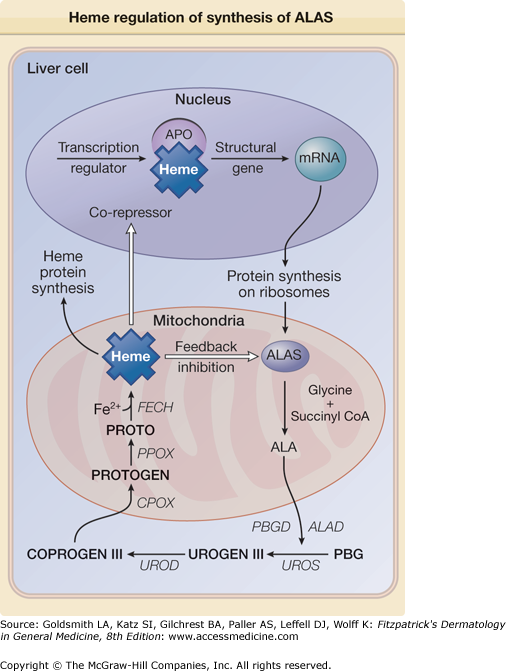
Heme is capable of regulating its synthesis by either directly inhibiting or repressing the synthesis of its rate-limiting enzyme ALAS. The repression may result from the binding of heme, as a corepressor, to an aporepressor protein (APO), which, when combined with heme, becomes a functional unit that blocks transcription of ALAS messenger RNA (mRNA). Heme may also block transport of the holoenzyme into the mitochondrion. ALA = δ-aminolevulinic acid; ALAD = δ-aminolevulinic acid dehydratase; CoA = coenzyme A; COPROGEN = coproporphyrinogen; PBGD = porphobilinogen deaminase; PROTO = protoporphyrin; PROTOGEN = protoporphyrinogen; UROGEN = uroporphyrinogen.
Figure 132-4
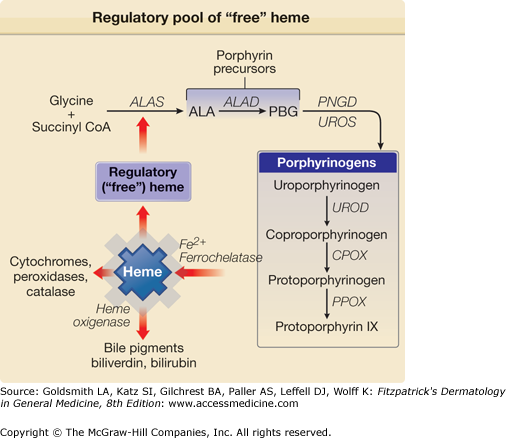
A regulatory pool of “free” heme unbound to apoproteins may be the critical determinant for regulation of synthesis of ALAS activity. ALA = δ-aminolevulinic acid; ALAD = δ-aminolevulinic acid dehydratase; CoA = coenzyme A; PBG = porphobilinogen; PBGD = porphobilinogen deaminase; COPROGEN = coproporphyrinogen; PROTOGEN = protoporphyrinogen; UROGEN = uroporphyrinogen.
|
Classification of the Porphyrias
Traditionally, most classifications of the porphyrias are based on the primary site of expression of the specific enzymatic defect, thereby distinguishing erythropoietic and hepatic types. From a dermatologic perspective, the porphyrias might also be classified into cutaneous and noncutaneous forms. However, from a general clinical perspective, we have chosen to classify the porphyrias into two subtypes: (1) nonacute and (2) acute thereby distinguishing patients in whom dermatologic findings predominate from those susceptible to potentially life-threatening acute neurovisceral attacks (Tables 132-3 and 132-4). We will employ this classification throughout this chapter. It should be pointed out that two of the acute porphyrias, variegate porphyria (VP) and hereditary coproporphyria (HCP), are also referred to as neurocutaneous porphyrias since patients with these diseases may have both neurovisceral and cutaneous manifestations.
Porphyria Cutanea Tarda | Erythropoietic Protoporphyria | Congenital Erythropoietic Porphyria | Hepatoerythropoietic Porphyria | |
|---|---|---|---|---|
Synonym(s) | Symptomatic porphyria, acquired hepatic porphyria, chemical porphyria | Erythropoietic porphyria, protoporphyria | Günther disease, congenital hematoporphyria, erythropoietic uroporphyria | Hepatoerythrocytic porphyria |
Inheritance | Autosomal dominant in approximately 20% of patients; otherwise acquired | Autosomal dominant with hypomorphic IVS3–48C allele; X-linked dominant; Autosomal recessive | Autosomal recessive | Autosomal recessive |
Age of onset | Usually in third or fourth decade; rare before puberty | Early childhood (1–4 years); late onset extremely rare | Usually infancy or first decade | Early infancy, before age 2 years |
Incidence | Most common porphyria worldwide | Second most common of the cutaneous porphyrias | Very rare (fewer than 250 cases) | Extremely rare (fewer than 40 cases) |
Photosensitivity | Moderate-to-severe with high interindividual variation; indistinguishable from variegate porphyria | Mild-to-severe; onset immediate (in minutes); interindividual variation | Severe to very severe | Severe |
Skin reactions (on sun-exposed areas) | Vesicles and bullae, erosions, crusts, milia, scarring, hypertrichosis; indistinguishable from variegate porphyria | Edema, erythema, urticarial plaques; very rarely bullae, skin thickening, waxy scars | Vesicles and bullae, erosions, hypertrichosis, hypermelanosis, mutilation | Vesicles and bullae, erosions, crusts, milia, scarring, hypertrichosis, mutilation |
Acute Intermittent Porphyria | Variegate Porphyria | Hereditary Coproporphyria | ALA Dehydratase Deficiency Porphyria | |
|---|---|---|---|---|
Synonym(s) | Swedish porphyria | Mixed porphyria, South African genetic porphyria, protocoproporphyria hereditaria, PCT hereditaria | Idiopathic coproporphyria | Plumboporphyria |
Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal recessive |
Age of onset | 10–40 years; extremely rare before puberty | Usually between ages 15 and 30 years | Any age | Early and late clinical onset has been described |
Incidence | 1.5 in 100,000; more common in Scandinavia and Lapland | Common in South Africa (3 in 1,000); relatively rare elsewhere | Rare (fewer than 50 cases reported) | None reported |
Photosensitivity | Absent | Moderate to severe with high interindividual variation; similar to that in PCT | Very rare | Absent |
Skin reactions (on sun-exposed areas) | None | Vesicles and bullae, erosions, crusts, milia, scarring, hypertrichosis; similar to those in PCT | Usually blistering | None |
An acute porphyric attack can present a constellation of symptoms and signs, among them, intense abdominal pain, vomiting, electrolyte dysregulation (hyponatremia), constipation, tachycardia, hypertension, muscle pain and weakness, seizures, paresis of the upper and lower extremities, paralysis, and a variety of other neurological and psychiatric symptoms.11 These myriad clinical manifestations inevitably mimic other diseases, thereby challenging the diagnostic acumen of even the most experienced clinicians particularly since the porphyrias are rare disorders.12 It is known that acute porphyric attacks can be precipitated by a variety of external factors, in particular porphyrinogenic drugs, ethanol, hormonal fluctuations, and diminished caloric intake from dieting or fasting.11,13 Yet in practice, it is often impossible to identify the immediate cause of an acute attack in an individual patient. Timely diagnosis of an acute porphyric attack can be lifesaving because several complications may have fatal consequences if not recognized and treated. It is crucial to keep in mind that many drugs are capable of inducing heme synthesis in the liver.11,13 Thus, in a patient an acute porphyria with episodic abdominal pain, treatment with certain analgesics may prolong or aggravate the attack. Diffuse abdominal pain can mimic acute appendicitis, diverticulitis, intestinal obstruction, or other painful gastroenterological disorders that may necessitate urgent surgical intervention.14,15 After administration of certain drugs (e.g., barbiturates) or during general anesthesia, acute attacks may ensue.1 A particularly problematic complication is muscle weakness. This can range from mild paralysis of small muscle groups to flaccid paralysis of multiple muscle groups resulting in paraplegia and respiratory compromise. These findings may be accompanied by severe sensory loss and amyotrophy.16 In some cases, paralysis may progress rapidly and can affect cranial nerves or develop into a Guillain–Barré-like syndrome.17
Nonacute and Acute Porphyrias
Patients with all forms of nonacute porphyria manifest cutaneous findings predominantly on sun-exposed body areas. The nonacute porphyrias include porphyria cutanea tarda (PCT), the most prevalent type of porphyria overall, hepatoerythropoietic porphyria (HEP), the homozygous variant of PCT, erythropoietic protoporphyria (EPP), XLDPP, the most recently recognized type of porphyria, and congenital erythropoietic porphyria (CEP) (Table 132-3). The nonacute porphyrias can present with variable cutaneous features, including mild-to-severe photosensitivity, increased skin fragility, vesicles and bullae, scarring with milia formation, burning and stinging, edema, pruritus, hypertrichosis, hyperpigmentation, and mild-to-severe scleroderma-like changes with tissue calcification.4
The acute porphyrias include acute intermittent porphyria (AIP), VP, HCP, and δ-aminolevulinic acid dehydratase (ALAD) deficiency porphyria (also known as plumboporphyria) (Table 132-4). Whereas AIP is the most prevalent form of acute porphyria worldwide, in some countries other acute forms may predominate, as is the case for VP in South Africa.1,2,18,19 Besides neurovisceral symptoms, individuals suffering from VP or HCP can also present with cutaneous findings, including increased photosensitivity, abnormal skin fragility, and bullous skin lesions on sun-exposed areas, erosions, chronic scarring, and postinflammatory pigmentary change. Thus, VP and HCP are also referred to as neurocutaneous porphyrias, to distinguish them from AIP and ALAD deficiency porphyria in which there are no cutaneous abnormalities.4,20
It is of interest that ethanol and estrogens, which can trigger acute porphyric attacks in AIP, VP, and HCP, can also exacerbate PCT, although acute attacks do not occur in this disease and the mechanisms likely differ.21
The most common cutaneous manifestation of the nonacute porphyrias is photosensitivity; it is observed in patients with several of these disorders (PCT, HEP, EPP, XLDPP, CEP) as well as in patients with VP and HCP who have both cutaneous and neurovisceral findings. The sine qua non of photosensitivity in porphyria is increased plasma and tissue porphyrins. Patients with AIP, in whom only nonphotosensitizing porphyrin precursors (ALA, PBG) are formed in substantially increased amounts, do not have photosensitivity nor do any patients with ALAD deficiency porphyria.
Patients with the erythropoietic porphyrias frequently complain of a painful and intense burning sensation and pruritus during or following sun exposure. A “priming” effect of sun exposure on cutaneous photosensitivity in EPP has been described in which a threshold exposure on one day that evokes virtually no reaction seems to augment the response to subsequent exposure on the following day.22 The cause of this is unknown, but the phenomenon suggests that repair of damage to skin by photosensitized porphyrins may be prolonged. These acute symptoms are notably absent in patients with the chronic hepatic porphyrias such as PCT and VP. Although there is some information about the pathophysiology of photosensitivity and sclerodermoid skin changes in the chronic hepatic porphyrias, the causes of the pigmentary alterations and hypertrichosis seen in these patients remain to be elucidated.
Porphyrins have certain unique photobiologic and spectroscopic properties that make them potent photosensitizers. Importantly, metal-chelated porphyrins (e.g., heme or FePROTO) show no fluorescence and are not photosensitizers. Porphyrins that are chelated to other paramagnetic metals, such as Mn2+, Co2+, or Zn2+ (e.g., chlorophyll, ZnPROTO), also do not fluoresce. For this reason patients with lead intoxication, in which ZnPROTO is elevated, do not have photosensitivity.23,24 However, metal-free porphyrins including, Uroporphyrin (URO), Coproporphyrin (COPRO), and Protoporphyrin (PROTO) in acidic solutions show a major absorption peak in the 400- to 410-nm region (Soret band); they also exhibit four additional absorption bands with decreasing intensity between 500 and 700 nm. Exposure of porphyrins to the Soret band spectra results in fluorescence emission peaks between 550 and 680 nm (Table 132-5). Although there is no single clearly defined pathway that can currently explain the photosensitization evoked by porphyrins and light, there are a number of potential cellular and soluble factors that are likely to be involved. Among them, reactive oxygen species (ROS) and their effect upon certain cells (erythrocytes, mast cells, polymorphonuclear cells, and fibroblasts), and soluble mediators (the complement system and matrix metalloproteinases) are discussed (Table 132-6). It is likely that a combination of these factors will prove to be responsible for the pathogenesis of the cutaneous lesions in the porphyrias.
Type | Peak Excitation (nm) | Peak Emission (nm) | Fluorescent Porphyrins |
|---|---|---|---|
Congenital erythropoietic porphyria | 398 | 619 | URO I, COPRO I |
Erythropoietic protoporphyria X-linked dominant Erythropoietic protoporphyria | 409 | 634 | FREE PROTO FREE and ZINC PROTO |
Porphyria cutanea tarda | 398 | 619 | URO I, III; COPRO III |
Variegate porphyria | 405 | 626 | COPRO III, PROTO |
Acute intermittent porphyria | 398 | 619 | URO III |
Hereditary coproporphyria | 398 | 619 | COPRO III |
Hepatoerythropoietic porphyria | 398 | 619 | URO I |
|
Of note, porphyrin abnormalities can also occur in lead poisoning, sideroblastic, hemolytic and iron deficiency anemia, renal failure, cholestasis, liver disease, and gastrointestinal hemorrhage. Of these, however, cutaneous photosensitivity has only been documented in rare cases of sideroblastic anemia.
Two acute porphyrias, AIP and ALAD deficiency porphyria, do not manifest cutaneous symptoms. In these diseases, the dysfunctional enzymes function early in heme biosynthesis and their substrates are nonphototoxic porphyrin precursors. However, patients with both AIP and ALAD deficiency porphyria as well as VP and HCP can manifest life-threatening acute neurovisceral attacks. Although the exact pathogenesis of these attacks is not well understood, it is known that the porphyrin precursors ALA and PBG are massively excreted from the liver during an acute attack and in particular ALA is known to be have neurotoxic properties. Furthermore, as a result of porphyria-related enzyme defects the net result may be heme deficiency in nerve tissue. Thus, the autonomic and peripheral nervous systems are particularly susceptible to porphyrin precursor induced neuropathy.25
(See Table 132-6)
Porphyrins such as URO, COPRO, and PROTO absorb light intensely around 400 nm and in the longer visible spectrum between 580 and 650 nm. Absorption results in the generation of “excited” state porphyrin molecules (Fig. 132-5). The initial excited state porphyrin generated has an extremely short half-life of less than 0.01 μs. Singlet excited state porphyrins may spontaneously convert to a triplet, another excited state that has a lower energy level but a longer half-life (in the order of microseconds to milliseconds). Because of their longer half-life, triplet-state porphyrins have a higher probability of reacting with biologic substrates and are likely candidates to mediate most porphyrin-associated photobiological reactions. Excited state porphyrin molecules ultimately must return to their normal ground states by releasing their absorbed energy in the form of light (fluorescence is emitted by singlet state molecules and phosphorescence by triplet states), heat, or by transferring energy to cell constituents (cell membranes, organelles, proteins, DNA) (Figs. 132-5 and 132-6).
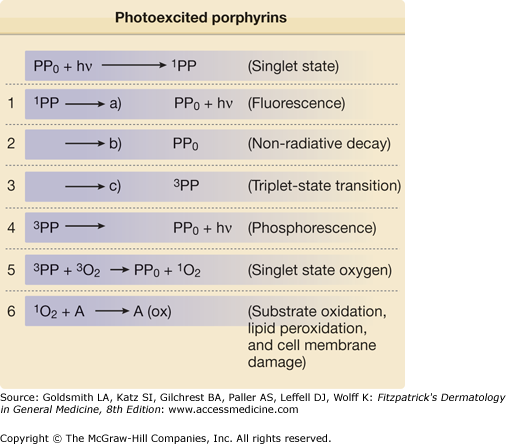
Figure 132-6
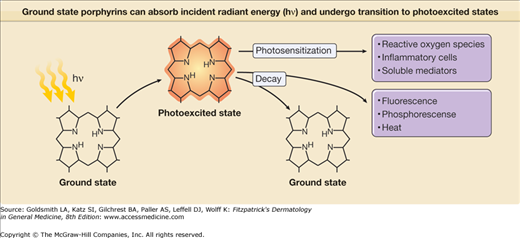
Ground state porphyrins can absorb incident radiant energy (hν), and undergo transition to photoexcited states. The photoexcited porphyrins generate reactive oxygen species, which damage cells by release of proinflammatory mediators; in turn, these may contribute to damaging reactions in biologic systems such as skin.
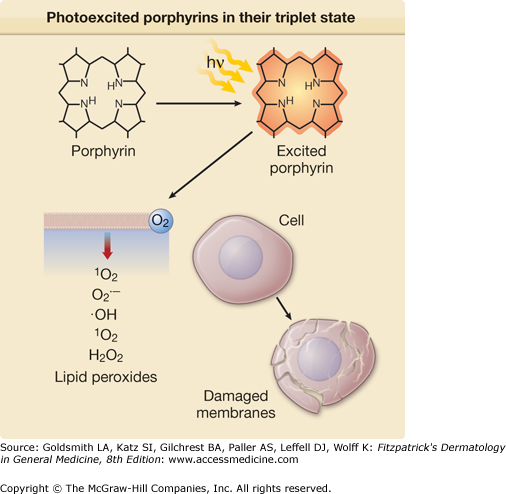
Excited porphyrins in their singlet and triplet states can also transfer their absorbed energy to oxygen molecules thereby creating reactive “excited” oxygen states.26 Cellular and tissue damage induced by photoactivated porphyrins is believed to occur primarily as a result of the formation of reactive singlet oxygen (1O2), as illustrated in Figs. 132-5 and 132-6.27,28 In some cases, the sensitized porphyrin may further react with oxygen to yield hydrogen peroxide (H2O2) or with water to form hydroxyl radicals (•OH). Those processes in which activated oxygen species play a role in photosensitization are referred to as photodynamic reactions.29 Most of the porphyrin-mediated cutaneous photosensitization reactions are essentially photodynamic (oxygen-dependent) and can be minimized or prevented if ROS are eliminated by inactivation processes known as quenching or scavenging.
Although these concepts regarding excited porphyrins and ROS are valid in simple systems and several hypotheses have been advanced regarding the mechanism of porphyrin-induced photosensitivity, their applicability to complex tissues such as the skin remains to be established.
Photoexcited PROTO elicits peroxidation of cholesterol groups in erythrocyte membranes, whereas URO or COPRO has no such effect. This difference is also true of mast cells, polymorphonuclear leukocytes (PML), and fibroblasts, and most likely relates to variations in lipid-water partitioning of the different porphyrins, which may in turn relate to their polarity. Lipophilic PROTO, a 2-carboxyl porphyrin, is more damaging to lipid-rich membranes as compared to the more hydrophilic COPRO and URO.30
Phototoxic damage to mast cells is associated with elevated serum histamine levels and dermal mast cell degranulation,31 and the phototoxic response can be suppressed by pretreatment with antihistamines (H1 blockers) or by intradermal injection of a mast cell secretagogue (compound 48/80).32
Mast cell degranulation occurs in the exposed skin of patients with EPP.33 Irradiation of PROTO-treated mast cells in vitro induces release of mast cell-derived mediators, although exposure to lower doses of PROTO and radiation results in inhibition of secretagogue-induced histamine release from these cells.34 In contrast, no mediator release is detectable when cells are radiated in the presence of URO. These findings may help to explain the differences in the clinical presentation of patients with EPP and PCT. The elevated PROTO in patients with EPP may induce the release of mast cell mediators in vivo following sun exposure, resulting in painful, pruritic erythema and edema. The absence of these changes in patients with PCT may be explained by the apparent inability of photoexcited URO to damage cutaneous mast cells.
Exposure of human PML to PROTO and radiation in vitro results in membrane damage, whereas photoexcited URO induces no such alterations.35 These results are strikingly similar to those obtained in studies with mast cells (see above). In an animal model, porphyrin-induced phototoxicity was associated with a dermal PML infiltrate31,36 and the response was markedly suppressed in leukopenic animals.32 These results appear to suggest that PML may be necessary but not sufficient to induce porphyrin-induced phototoxicity.
Differential phototoxic effects of various porphyrins have also been verified in studies using dermal fibroblasts. An increase in collagen biosynthesis is observed following incubation of fibroblasts with URO.37 This effect is independent of irradiation and may partly explain the sclerodermoid changes observed in patients with PCT, which can occur in sun-exposed and in sun-protected areas. In contrast, PROTO induces photolysis of fibroblasts in vitro.38,39
Photoexcited URO has been shown to coordinately induce interstitial collagenase (MMP-1), 72-kDa type IV collagenase (MMP-2) and stromelysin-1 (MMP-3) in human dermal fibroblasts in vitro.40 The induction of the MMPs by porphyrin photosensitization suggests that activation/release of these enzymes could contribute to the blistering process.
The complement system is activated by photoexcited porphyrins such as URO and PROTO. In vitro exposure to Soret band radiation activates the complement cascade and releases anaphylatoxins.41,42 Exposure of the skin of patients with EPP and PCT to Soret band energy in vivo also generates anaphylatoxins.43 Granular and homogeneous deposits of the complement membrane attack complex C5b9 have been observed in skin biopsies obtained from the skin of patients with PCT.44 It is of interest that target tissue heme pathway enzymes may be a crucial determinant of skin susceptibility to porphyrin photosensitization. Using a mouse model of EPP it has been shown that in transplanting bone marrow from EPP animals to wild-type mice the skin of the recipients was resistant to photosensitivity despite very high levels of plasma porphyrins suggesting that the normal FECH levels in the recipients somehow protected them against phototoxicity.45
In summary, cutaneous phototoxicity in the porphyrias is directly related to the interaction of porphyrins with light of the Soret and other bands in the solar spectrum, resulting in the generation of ROS. This in turn can induce lipid peroxidation and cell membrane alterations. Release of mediators and enzymes from cells such as mast cells and PML contributes to the inflammatory response. Variations in solubility influenced by the lipid–water partitioning of porphyrins may account for the striking patterns of the skin findings observed in various types of porphyria. It is important to emphasize that porphyrinogens (reduced porphyrins) are the true intermediates in heme synthesis. The irreversibly oxidized porphyrins, with the exception of PROTO, do not function as substrates for the enzymes of the pathway. Thus, porphyrins are actually heme pathway by-products, which are of special interest to dermatologists because of their unique photosensitizing properties.
The Nonacute Porphyrias
|
PCT occurs throughout the world and is the most common of all the porphyrias.46 The prevalence is estimated at 1:10,000.47 The disease most often begins in middle-aged individuals but can develop earlier. Prior to the widespread use of oral contraceptives, PCT developed predominantly in males.48,49 In contrast, others have emphasized that the sex incidence is approximately equal.50 The rising incidence of PCT in females is probably due to the widespread ingestion of estrogens in oral contraceptives or in hormone supplements. It should be noted that males treated with estrogens, for example, as adjunctive therapy for carcinoma of the prostate, have also developed PCT.
PCT is due to either an inherited or acquired deficiency of UROD, the fifth enzyme in the heme pathway (Figs. 132-1 and 132-2) and it has a molecular mass of about 42 kDa.51 UROGEN I and III each contain eight carboxyl groups as side chains, four of which are acetate (–CH2COOH) and four of which are propionate (–CH2–CH2–COOH) moieties (Fig. 132-2). The soluble cytosolic enzyme UROD catalyzes the sequential oxidative decarboxylation of the four carboxyl groups of the acetate side chains to methyl groups to form COPROGEN (Fig. 132-2). Decarboxylation first occurs on ring D, after which the enzyme turns around to decarboxylate rings A, B, and C in a clockwise fashion. This converts the original 8-carboxyl porphyrinogen (UROGEN I or III) first to 7-carboxyl, then to 6-carboxyl, and 5-carboxyl porphyrinogen. The 5-carboxyl porphyrinogen can then undergo decarboxylation of its last acetyl group to form the 4-carboxyl porphyrinogen that is known as coproporphyrinogen (COPROGEN I or III) (Fig. 132-2). These intermediates are also referred to as hepta-, hexa-, penta-, and tetracarboxylate porphyrinogens. COPROGEN I cannot be further metabolized to heme.
Human UROD is encoded by a single copy of the gene of the same name that is located on chromosome 1p34 spreads over a genomic distance of 3 kb, and contains ten exons.51 The human UROD cDNA has been isolated, and the deduced sequence was found to be equivalent to 367 amino acids, consistent with the molecular mass and the amino acid composition of the purified protein.
Most classifications of PCT separate the disorder into at least two broad categories, both associated with decreased UROD activity: (1) acquired PCT, also referred to as sporadic or type I PCT; and (2) hereditary PCT, also referred to as familial or type II PCT.
In acquired (type I) PCT, the enzyme is deficient only in the liver and no mutations have been detected in the coding or promoter sequences of the UROD gene (Table 132-2).46,52 Some of the precipitating substances (e.g., alcohol and estrogen) may provoke PCT only in selected individuals and others [e.g., hexachlorobenzene (HCB)] in practically all exposed individuals (Table 132-7).
|
Hereditary (type II) PCT is an autosomal dominant disorder and the residual UROD activity is decreased approximately 50% in all tissues, including RBC and cultured skin fibroblasts.53–56 Decreased enzyme concentration appears to follow a bimodal distribution, suggesting two overlapping groups of patients: a large group (>80%) with normal UROD concentration and a small group (<20%) in who the amount of detectable enzyme is about half normal. Some patients with type II PCT were found to have UROD activity at the lower end of the normal range. The clinical penetrance of type II PCT is relatively low (approximately 20%), so that the majority of individuals with the inherited enzyme defect do not manifest the disease, suggesting that additional genetic or nongenetic factors are needed for disease expression. To date, more than 50 mutations in the UROD gene have been identified in type II PCT, mostly in exons 5–10, which encode 75% of the protein that either decrease the stability of the enzyme or produce defective pre-mRNA splicing.3,46,47,51,56–59
It is important to emphasize that not all patients with a positive family history of PCT will have type II disease. In support of this notion, several patients have been described with one or more relatives with type I PCT but with normal RBC UROD activities.60 This latter category of PCT has been designated as type III by some investigators.54 These patients could either have inherited some form of UROD that is immunochemically indistinguishable from the normal enzyme but which is uniquely susceptible to inhibition in the liver, or they may have a second inherited enzyme deficiency unrelated to UROD. These possibilities require further investigation.
It is likely that some patients who were initially reported as having hereditary PCT actually had VP, another dominantly inherited porphyria. PCT and VP may occur in different members of the same family, in the so-called dual porphyrias.61–63 Another form of dual porphyria in which PCT and AIP coexist has also been described.64
Numerous agents are known to contribute to the development of PCT (type I, acquired, or sporadic) including alcohol, estrogens, iron, viral infections (hepatitis C and HIV), polychlorinated hydrocarbons, particularly HCB and TCDD, and hemodialysis in patients with renal failure (Table 132-7). Each of these precipitating factors is discussed briefly below.
Alcohol ingestion has long been recognized to exacerbate PCT. Ethanol has been shown to induce hepatic ALAS1 in patients with PCT.65 Erythrocyte UROD activity is diminished in healthy subjects following acute ethanol ingestion and in chronic alcoholics.66 Ethanol can also inhibit the activity of other enzymes in the heme pathway, including FECH and ALAD. Chronic alcoholism leads to suppression of erythropoiesis67 and increased absorption of dietary iron, perhaps linked to inherited mutations associated with hemochromatosis (see below). The fact that ALAS1 is increased in patients with hepatic cirrhosis without porphyria raises questions concerning the relevance of alcohol effects on ALAS1 in the clinical expression of PCT.68
The widespread use of estrogens as contraceptive agents or as hormone supplements for postmenopausal replacement therapy in females and as adjunctive hormonal therapy in males with prostatic carcinoma has been associated with PCT.50 The mechanism of the estrogen effect on the expression of PCT has not been elucidated. Although diethylstilbestrol, an estrogen, induces hepatic ALAS1,69 this would not explain the distinctive porphyrin excretion pattern found in PCT. The vast majority of patients receiving estrogens do not manifest the biochemical abnormalities associated with PCT.
This fungicide caused an “epidemic” of a PCT-like syndrome in Southeastern Turkey in the 1950s.70 It was added as a preservative to wheat intended for planting, but, because of a famine, several thousand individuals of diverse ethnic origin, mostly children, ingested the seed wheat and subsequently developed typical PCT. Over 4,000 cases of this syndrome were reported from 1956 to 1961. The porphyrin excretion pattern and the cutaneous findings in these patients were quite similar to those seen in PCT evoked by ethanol or estrogens. The outbreak of PCT in Turkey caused by ingestion of HCB indicated that the disease could occur in nongenetically predisposed individuals.
Twenty-five years later, the most common clinical findings in these HCB-poisoned individuals were those of chronic porphyria, including sclerodermoid scarring (84%), hyperpigmentation (78%), hirsutism (49%), thyromegaly, and increased skin fragility (38%).71 A painless arthritis was seen in two-thirds of affected individuals, and a variety of neurologic signs and symptoms occurred in the majority. Stool and urine porphyrins remained elevated in many patients.
Studies have shown that the chronic administration of HCB to experimental animals produces excessive porphyrin accumulation in the liver in a pattern quite similar to that seen in PCT in humans.72 These data are consistent with the hypothesis that chlorinated hydrocarbons, such as HCB, or their metabolites inhibit hepatic UROD, leading to excessive hepatic storage of URO and other acetate-substituted porphyrins.73,74 Experimental studies have also shown that HCB can inactivate UROD, thereby abolishing catalytic activity without changing the amount of immunoreactive enzyme protein.75 Chemical porphyria, similar to PCT, is caused by other chlorinated hydrocarbons such as the polychlorinated biphenyls (PCBs) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a by-product in the synthesis of the herbicide 2,4,5-trichlorophenoxyacetic acid.76 Additional studies of the porphyrinogenic effects of chlorinated hydrocarbons suggest that metabolic activation of the compounds mediated by cytochrome P450 1A2 and involving iron-generated ROS is associated with an attack on the catalytic site of UROD.56,77
TCDD is a toxic environmental pollutant chemical. Among its numerous effects are chloracne, liver damage, and hepatic porphyria in experimental animals and perhaps also in humans.78 It has been shown that the hepatic porphyrinogenic effect of TCDD can be abolished in mice by first depleting them of iron.79 Furthermore, it is known that highly inbred mouse strains vary in their susceptibility to induction of hepatic porphyria by TCDD, indicating that the porphyrogenic effect of this hydrocarbon is modulated by as yet undefined genetic factors.80
Serum iron and ferritin concentrations are often elevated or in the upper range of normal in PCT, confirming the important role of iron in the pathogenesis of the disease.81 Hepcidin is a 25-amino acid peptide that is encoded in humans by the HAMP gene and it is secreted by the liver and inhibits iron transport by binding to the iron channel ferroportin located on the surface of gastrointestinal enterocytes and the plasma membrane of reticulo-endothelial cells. The net result is that iron is trapped in these cells and cannot be absorbed. Ajioka et al showed that hepatic HAMP expression is decreased in patients with PCT suggesting that the hepatic siderosis associated with PCT likely results from dysregulated HAMP.82 Hepatic iron overload accompanies clinical PCT in practically all cases, and elevation of plasma iron is found in one-third to one-half of patients.50 In PCT, the quantity of iron that can be mobilized by phlebotomy indicates that total body iron stores are approximately twice normal. Ferrokinetic studies in patients with PCT are said to be normal. The long remissions that follow repeated phlebotomy and the apparent ineffectiveness of this treatment if supplemental iron is administered concomitantly suggest that iron plays a role in the excessive hepatic porphyrin production of PCT.83 PCT is particularly common where alcoholism and iron overload occur together.
The role of iron in the pathogenesis of PCT is complex, and several hypotheses have been proposed to explain it. Iron may directly inhibit UROD. However, studies with purified UROD prepared from human erythrocytes show that the purified enzyme is not inhibited by Fe2+ or Fe3+.84 Chronic iron overload can produce peroxidative damage to lipid-rich mitochondrial and microsomal membranes in the liver of experimental animals, but the relationship of this toxic effect to changes in hepatic heme synthesis has not been clearly defined.85 An increased frequency of the hemochromatosis (HFE) gene C282Y mutation has been found in British patients with sporadic PCT.86,87 This mutation is responsible for much of the iron overload in populations of European descent. A second mutation in the HFE gene, H63D, may also be associated with PCT in some populations.88 PCT is rare in heterozygotes for each of these mutations but 20% of British patients with PCT are C282Y homozygotes. C282Y homozygosity is an important susceptibility factor for both type I and type II PCT.57 However, iron stores are increased in many patients with PCT without mutations in the HFE gene.46
Iron may have a permissive effect on the inhibition of UROD by halogenated hydrocarbons, and it can also enhance the induction response of hepatic ALAS1 to drugs.89,90 Although such an iron-augmented increase in ALAS1 activity could lead to enhanced porphyrinogenesis, this alone would not explain the porphyrin excretion pattern seen in PCT. It is known that addition of ferrous iron to liver in vitro causes a marked increase in porphyrin synthesis and inhibits UROS activity, providing an explanation for the URO isomer I excess characteristic of PCT.91 A mouse model of type II PCT has been described in which one allele of UROD is disrupted and the animals bred to mice null for HFE thereby creating a PCT-like phenotype.92
There is an association between PCT and hepatitis C virus (HCV) infections and combined HCV and HIV infections.56,93,94 The role of these viruses in the pathogenesis of PCT is unclear, although some connection with the HFE gene mutation H63D has been suggested. It is also possible that the connection is fortuitous and secondary to nonspecific hepatotoxic effects of these viruses.
From these known effects of alcohol, estrogens, chlorinated hydrocarbons, iron, and viral infections on the heme pathway, it is clear that each of these could contribute to the excessive hepatic porphyrinogenesis characteristic of PCT.56 There is growing evidence that hepatic siderosis is the critical pathologic endpoint in PCT and that the other agents somehow intensify the ability of iron to attack the catalytic site of UROD. The clinical expression of PCT is therefore dependent upon the interaction of a number of factors, both genetic and environmental. However, it is important to note that the ingestion of drugs that have been associated with inducing acute neurovisceral attacks in the acute porphyrias is not known to exacerbate similar neurological crises in PCT.
Vesicles and bullae followed by erosions and crusting occur predominantly in areas subject to repeated trauma (Figs. 132-7 and 132-8). There is increased skin fragility, usually on the dorsa of the hands (Fig. 132-8). The traumatized skin becomes crusted and, as the lesions resolve, areas of scarring may ensue. Numerous small milia can develop, particularly on the fingers and hands. These pearly white to yellow papules occur in each of the hepatic porphyrias with cutaneous photosensitivity (PCT, VP, and HCP) (Fig. 132-7). Although the cutaneous lesions are primarily seen on the light-exposed areas, patients are often unaware that sunlight plays a role in producing their lesions because the acute painful and burning photosensitivity, so characteristic of the erythropoietic porphyrias, is rare in PCT. However, most patients do recognize that their skin condition worsens in the spring and summer and seems to improve in the fall and winter.
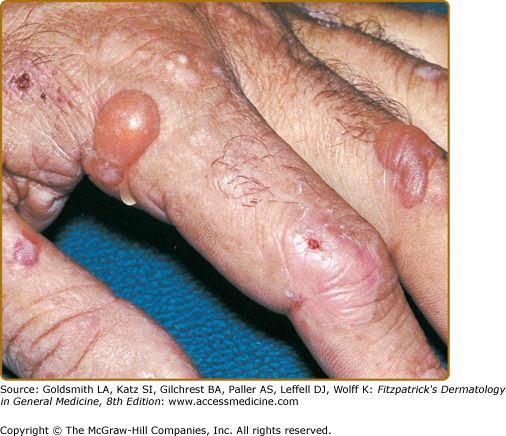
Other skin changes seen in PCT include hyperpigmentation and hypopigmentation that may be mottled, resembling chloasma. There may be an associated purplish-red (“heliotrope”) suffusion of the central part of the face, particularly involving the periorbital areas, which may bear a striking resemblance to the plethora seen in polycythemia rubra vera (Fig. 132-9). This is not seen in the porphyrias of bone marrow origin.
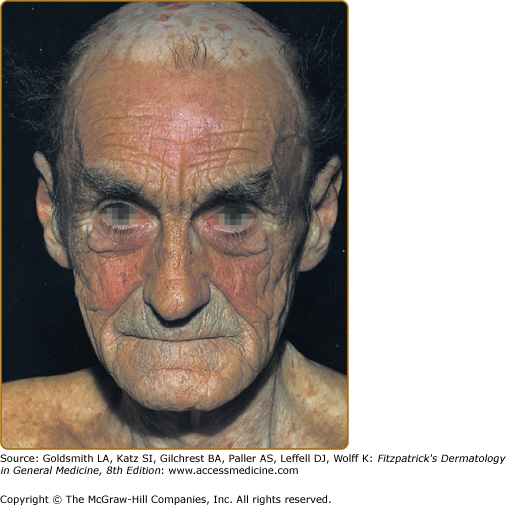
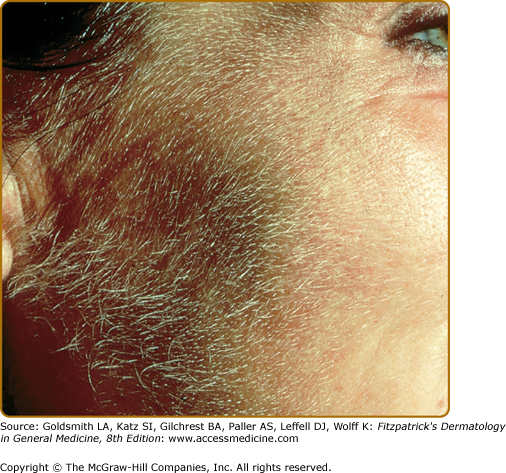
Hypertrichosis (nonvirilizing) is a useful diagnostic sign that often brings the female patient to the dermatologist (Fig. 132-10). Facial hypertrichosis develops gradually and occurs in both males and females. The hair may vary in texture between fine and coarse and in color between light and dark. These hairs are particularly prominent along the temples and the cheeks, but may occasionally involve the trunk and extremities in severe cases. Such hair may continue to grow, darken, and thicken, particularly on the cheeks, the forehead between the eyes, and at the hairline of the scalp. Males may complain that shaving is more difficult and that the growth pattern of their beard has changed. A particularly severe form of hypertrichosis may occur in younger children with PCT and HEP. In the reports of HCB poisoning in Turkey, some of the children were described as “monkey-like” because of marked hypertrichosis.95 The mechanism of this phenomenon is unknown; androgen levels are reported to be normal. It is possible that surface receptors or growth factors for hair bulb keratinocytes are activated by the dual action of light and porphyrins. The hypertrichosis of PCT usually improves slowly following depletion of excessive hepatic iron stores.
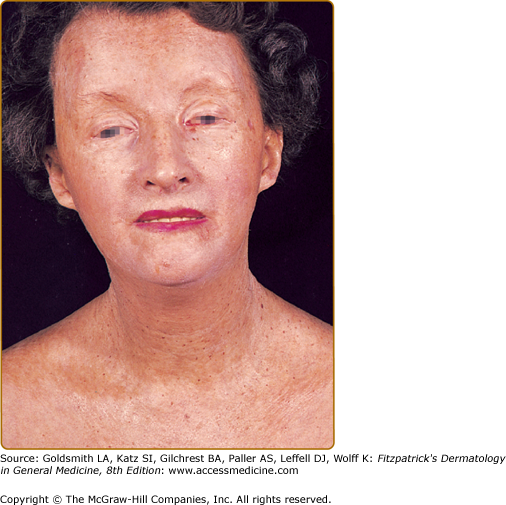
Sclerodermoid plaques can occur in PCT and in HEP and typically develop on both light-exposed and light-protected body areas (Figs. 132-11A and 11B). These are usually scattered, waxy yellow to white, indurated plaques that closely resemble, both clinically and histopathologically, morphea or scleroderma. There is some evidence to indicate that PCT may occur concomitantly with true scleroderma but this seems to be quite rare. As discussed earlier, URO I stimulates collagen synthesis in human skin fibroblasts.37 In some patients, calcification has developed in these sclerodermoid plaques, necessitating surgical excision and grafting.
PCT-like syndromes are occasionally seen in association with hepatic tumors and lupus erythematosus.96,97 Subepidermal bullous dermatoses mimicking PCT clinically and histologically have been described (see Section “Pseudoporphyria”). A number of cases of true PCT have occurred in patients with renal failure undergoing hemodialysis.98 Confirmation of the diagnosis rests on detection of markedly elevated plasma porphyrins (usually 5–100-fold) and increased ISOCOPRO in the feces.
Patients with PCT excrete increased amounts of porphyrins in the urine, which rarely may exhibit characteristic pink–red fluorescence when examined with a Wood’s lamp. The porphyrin excretion pattern of PCT has three main features: (1) increased urinary excretion of URO and of other acetate-substituted porphyrins; (2) a distinctive pattern of excretion of isomer series I and III porphyrins; and (3) increased excretion of fecal ISOCOPRO.56,99,100 PCT patients excrete greatly increased amounts of urinary 8-carboxyl URO and also porphyrins with 7-, 6-, and 5-carboxyl groups; 4-carboxyl porphyrin (COPRO) is also increased but to a lesser extent than URO and rarely surpasses 600 μg per 24 hours (Table 132-8). In PCT, the hepatic UROD deficiency results in the accumulation of 5-carboxylporphyrinogen III (Figs. 132-1 and 132-2). This can be utilized as a substrate by the enzyme CPOX and thereby forming dehydroisocoproporphyrinogen, which in turn is oxidized to ISOCOPRO, resulting in the characteristic elevation of this compound in the feces of these patients.
Blood | Stool | Urine | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Type of Porphyria | RBC URO | RBC COPRO | RBC PROTO | Plasma | URO | COPRO | PROTO | Color | ALA | PBG | URO | COPRO |
Nonacute | ||||||||||||
Porphyria cutanea tarda | N | N | N | ↑URO | ++ | + ISOCOPRO |
| Pink to red | N | N | ++++ | ++ |
Hepatoerythropoietic porphyria | N | ± | ++++ | ↑URO | N |
| N | Pink to red | +++ | ISOCOPRO | ||
Erythropoietic protoporphyria | N | N to + |
| ↑PROTO | N | ++ |
| N | N | N | N | N |
X-linked dominant protoporphyria | N | N to + |
| ↑PROTO | N | ++ |
| N | N | N | N | N |
Congenital erythropoietic porphyria |
|
| +++ | ↑URO & COPRO | + | +++ | + |
| N | N | ++++a | ++a |
Acute | ||||||||||||
Acute intermittent porphyria | ||||||||||||
Latent | N | N | N | N | N to + | N to + | N to + | Red to purple |
|
| N | N to + |
Acute | N | N | N | N | N to + | N to + | N to + | Red to purple | ++ to ++++ | ++ to ++++ | ++++ | ++ |
Variegate porphyria | ||||||||||||
Latent | N | N | N | ?N | N | +++ | ++++ | N | N | N | N | N |
Acute | N | N | N | ?N | ++ | ++ | +++ | Pink to red |
|
| +++ | +++ |
Hereditary coproporphyria | ||||||||||||
Latent | N | N | N | ?N | ++ |
| N to + | N | N to + | N to + | N | N |
Acute | N | N | N | ?N | + |
| N to + | Red | ++ to N | ++ to N | ++ | +++ |
ALA dehydratase deficiency porphyria | N | N | ++ | ↑ALA ↑COPRO ↑PROTO | N | + | + | ? |
| N | + | ++ |














The 8-carboxyl URO and 7-carboxyl porphyrins are the predominant urinary porphyrins in PCT (>90% of total porphyrins). The urinary porphyrin excretion pattern is a mixture of type I and type III isomers. URO is about 60% type I isomer and 40% type III; the 7- and 6-carboxyl porphyrins are >90% type III and <10% type I isomer; the 5- and 4-carboxyl porphyrins are about 50% each isomer. This distinctive isomer pattern is found consistently in patients with PCT.
In general, only trace amounts of URO are present in the stools of normal individuals. The porphyrin content of stool is increased in PCT and consists primarily of ISOCOPRO (type III), 7-carboxyl porphyrin, and lesser amounts of URO and COPRO. The total daily 24-hour fecal porphyrin excretion may exceed total urinary porphyrin excretion.
The ratio of URO to COPRO in the urine is often helpful in differentiating PCT and VP. Thus, in PCT, the URO:COPRO ratio usually exceeds >3:1, whereas in VP the ratio is typically less than 1:1. Occasionally, 24-hour urine porphyrins will be normal or only slightly increased in a patient with the cutaneous findings of PCT. This should alert the physician to suspect the diagnosis of VP and thus to evaluate stool porphyrins, as these are almost always elevated in patients with VP (see Section “Variegate Porphyria”).
It should be emphasized that in patients with clinical signs of PCT a fluorescent screening test for urinary porphyrins is often negative. In such patients, it is absolutely essential to measure plasma porphyrin levels and perform quantitative 24-hour urine URO and COPRO determinations and quantitative stool PROTO and COPRO determinations preferably using high-performance liquid chromatography, which often permit differentiation of PCT from VP. Rarely, some patients with VP will have normal fecal porphyrin excretion, and bile porphyrin measurements may be helpful in evaluating such patients.101
Virtually all patients with PCT have excessive total body iron stores manifested as increased serum iron, ferritin, and/or hepatocellular iron.46,56 Occasionally, there is mild erythrocytosis. An abnormal glucose tolerance test occurs in a minority of patients.
Biochemical tests for liver function often reveal elevated serum transaminases and γ-glutamyltranspeptidase levels. Serum iron and ferritin concentrations may be elevated. The measurement of erythrocyte UROD is useful for the detection of patients with type II PCT. Mutational analysis is an essential part of evaluating these patients (see below).
The characteristic histopathologic finding in PCT is a subepidermal blister (Fig. 132-12). Bullae characteristically show a corrugated, undulating base that has been termed festooned.102 There is little or no inflammatory infiltrate. PAS stain reveals a mild degree of thickening of the papillary vessel wall, not nearly so marked as that seen in patients with EPP. Reticulin staining demonstrates slight proliferation of fibers along the basement membrane. Direct immunofluorescence studies reveal deposition of C3, C5b-9, and IgG in a granular pattern at the dermal–epidermal junction and in and around vessel walls in affected individuals.44,103 These changes are most apparent in sun-exposed areas of patients with active disease and high urinary porphyrin excretion, and they decrease substantially in patients after appropriate treatment. It is possible that the deposition of immunoglobulins and complement is a nonspecific result of injury to the cutaneous tissue. Damage of the upper dermal vessels and at the dermal–epidermal junction suggests that structural changes evoked by porphyrin photosensitivity may be responsible for the unique skin fragility seen in PCT.
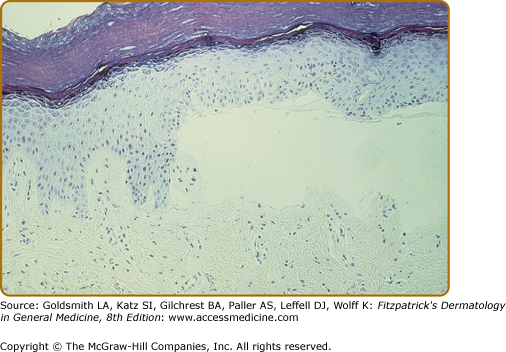
Regardless of these findings, it should be emphasized that histopathologic examination does not substantially contribute to confirming the diagnosis of PCT. This is also true for the other cutaneous porphyrias and, consequently, it is not essential to obtain a skin biopsy if one of the cutaneous porphyrias is suspected, mainly for two reasons. First, simple noninvasive biochemical laboratory techniques (see above) can usually permit presumptive diagnosis of porphyria and, second, external trauma (such as a biopsy or excision) inevitably constitutes an avoidable risk for delayed and/or dysfunctional wound healing that is a characteristic feature of all cutaneous porphyrias.
Other dermatoses that can be confused with PCT include VP, HCP, mild forms of HEP and/or CEP, pseudoporphyria, scleroderma, and epidermolysis bullosa acquisita (EBA). Each of these can be differentiated by appropriate porphyrin studies. Careful evaluation of urine, stool, and plasma porphyrins will almost always permit confirmation of the diagnosis of PCT. Nonsteroidal anti-inflammatory drugs such as naproxen and antibiotics such as the tetracyclines and nalidixic acid as well as a variety of other agents may rarely produce bullous lesions closely resembling PCT but in contrast show no evidence of abnormal porphyrins (see Section “Pseudoporphyria”).
(See Box 132-1). Initially, a careful history should be taken in an effort to identify an environmental toxin, for example, alcohol, estrogen, or chlorinated hydrocarbon, which may have triggered the disease.46,56 If HCV or HIV infection is present these should be managed appropriately. Elimination of toxin exposure alone may result in gradual improvement. However, in most patients with PCT, more aggressive treatment is usually appropriate to accelerate therapeutic benefit and this currently consists of either repeated phlebotomy56,104,105 or orally administered antimalarial drugs, either chloroquine or hydroxychloroquine106,107 or a combination of both.108 Other forms of treatment that have been described include administration of iron chelators109,110 and the oral administration of cholestyramine.111
Type | Management |
|---|---|
Porphyria cutanea tarda |








