Introduction
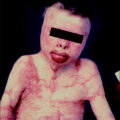
A large burn is not a simple injury, but a very complicated disease. This statement, restated from its publication in 1840, holds true with additional force in 2025. Massive destruction of skin tissue by burns stimulates many complex reactions that are still only partly understood. Malfunction of every organ system complicates the responses of patients to large burns. These malfunctions can be clarified by examination of the body after death. Postmortem examination also may reveal unsuspected infections or adverse effects of therapy. In addition, postmortem examination leads to review of the circumstances of injury and of the causal sequence in which complications occurred. Analysis of an entire case from the point of view of pathogenesis often clarifies the nature of the patient’s most significant problems. The manner of death is accident in most cases, but collection of additional information on the circumstances of the initial injury sometimes reveals evidence of homicide. After presentation of the major systemic problems that occur in burn patients, this chapter reviews some of the observations made at autopsy organized by organ system. It also surveys experimental evidence that bears on pathogenic mechanisms relevant to disease processes seen at autopsy. The observations reviewed here are drawn from the experience of more than 300 autopsies performed on burned children who died at the Shriners Hospital Children’s Texas in Galveston, Texas, from 1971 to the present.
Systemic reactions to burns
Hypoxia and ischemia
Immediately after burn injury, massive loss of intravascular fluid into the burned tissue begins to occur. , Unless this fluid loss is replaced very promptly and carefully, serious hypovolemia develops. Hypovolemia and the resulting reduction of tissue perfusion to levels less than that necessary for cellular survival, which is defined as ischemia, causes necrosis of certain cells. Typically, the cells first affected by ischemia are those with the greatest oxygen demand, including neurons in the brain, cardiac myocytes, intestinal epithelial cells, and proximal tubular epithelial cells in the kidney. After the death of selected cell types or whole segments of organs (infarcts), responses are generated that often lead to further injury of remote organs. Cellular necrosis stimulates an intense acute inflammatory reaction. In the skin, the tissues at the base of the zone of necrosis become acutely inflamed. Cytokines released by inflammatory cells and surviving cells in the tissue have effects throughout the body. In addition, in endothelial cells injured by hypoxia, the enzyme xanthine dehydrogenase is converted to xanthine oxidase, which releases superoxide during degradation of adenosine, which in turn is released by necrotic cells. , Superoxide, released into the circulation by this metabolic process and by neutrophils, can injure the lung by damaging both endothelial and epithelial cells and allowing protein-rich fluid to exude into alveoli. In experimental models of burn injury, as well as in models of ischemia-reperfusion injury, the lungs have been shown to be injured by these processes. , Ischemia as a result of reduced perfusion may lead to necrosis of pancreatic acinar epithelium and acute pancreatitis. Thermal injury to skeletal muscle, or lack of perfusion of muscle, may lead to local exudation of fluid and development of such high pressures in fascial compartments that arterial perfusion is prevented. This compartment syndrome, unless relieved by prompt surgical intervention, leads to necrosis of muscle throughout the compartment. The consequences of massive necrosis of muscle often include secondary injury to the lungs, owing to release of reactive oxygen species, and myoglobinuria with secondary renal damage. At the time of injury, patients frequently inhale sufficient carbon monoxide, which compromises the oxygen-carrying capacity of the blood. The resultant tissue hypoxia can cause death at the scene, and if the patient survives it can be sufficient to lead to irreversible neuronal injury, cerebral edema, and brain death. Hypoxemia, sometimes related to carbon monoxide intoxication, also contributes to cardiac and renal injury. In addition, when fires occur in closed spaces, the flashover process consumes all available oxygen so that the patient’s environment may contain too little oxygen to sustain life. Occasionally a burn victim is found without pulse or respiratory effort, probably as a consequence of hypoxia, and is revived by cardiopulmonary resuscitation. In such cases ischemic and hypoxic injury may be profound in multiple organs.
Infection
Necrotic skin provides an excellent environment for proliferation of bacteria and fungi, and as long as necrotic tissue remains, the risk of infection remains high. Immunosuppression contributes to this risk, and burn patients develop serious infections with agents usually encountered only in patients treated with immunosuppressive drugs. The mechanisms of this immunosuppression are still under investigation, but they include excessive secretion of glucocorticoids, abnormal cytokine signaling, and altered maturation of neutrophils and macrophages. When burn wounds become infected and large numbers of bacteria accumulate, those with high pathogenic capacity invade the adjacent viable tissue, produce further necrosis, and gain access to the circulation. This is the condition of burn wound sepsis, which historically has been the leading cause of death in burn patients. In Linares’s series of 115 autopsies, sepsis was present in 73%, as documented by positive blood culture and demonstration of invasive infection of viable tissue. , When pneumonia was found at autopsy in such a case, the cause of death was classified as burn wound sepsis, with pneumonia as a contributing cause. In recent years, the autopsy reports have listed infection of the lungs as the immediate cause of death, with burn wound sepsis listed as a contributing cause when the same diagnostic criteria were fulfilled. The basis for this interpretation is that in cases of pulmonary infection following septicemia the lungs are often several times the normal weight and were so filled with edema and hemorrhage that there was little remaining capacity for air, and clinical records consistently showed that progressive hypoxemia occurred during the hours before death. In 80% of these fatal cases of sepsis, the burn wound was the source of the infection. In the remaining 20%, the source of septicemia was unclear, but infection, ulceration, and necrosis were frequently found in the larynx, trachea, or bronchi. The systemic veins draining the trachea can carry bacteria through the right ventricle into the pulmonary arteries, as do the veins in infected wounds. The veins draining the bronchi drain directly into pulmonary arteries. The most important pathogens in cases of lung infection following invasive infection of burn wounds are Pseudomonas aeruginosa, Acinetobacter baumannii/haemolyticus, Klebsiella pneumoniae, Escherichia coli, and Enterobacter species, all gram-negative bacteria, with P. aeruginosa accounting for the great majority of cases.
It is important to assess the presence and extent of infection within the burn wound, both by careful clinical examination of wounds and by biopsy of suspicious areas. A high index of suspicion serves the burn patient well. All biopsy and excision specimens in our institution are sampled and studied histologically with special stains (a tissue Gram stain for bacteria and methenamine silver for fungi). In large excision specimens, samples are taken from sites of especially deep tissue injury and sites that show abnormal discoloration of dermal or subcutaneous tissue. When infectious microorganisms are found, it is important to determine their location with respect to the boundary between living and necrotic tissue. This boundary may be irregular, but it is generally distinct and marked by inflammation in wounds several days old, but may be indistinct in very fresh specimens, as karyolysis takes some time to develop in burn wounds. Wound infections generally begin with colonization of the skin surface and proliferation of organisms on the surface, often with extension into hair follicles, followed by growth within the necrotic tissue. Both the coagulum on the surface and the necrotic epidermis and dermis are considered part of the burn eschar. Growth within necrotic tissue is considered evidence of invasion of necrotic tissue, however, and potentially more dangerous than growth on the surface of necrotic skin, under a layer of fibrin, and debris. Even when quantitative cultures show more than 10 5 bacteria per gram of tissue, when careful histologic study shows that the organisms are limited to the skin surface or the superficial necrotic tissue, the risk of sepsis appears to be low. Such growth on or in necrotic tissue, however, sets the stage for invasion of viable tissue. The finding of clusters of bacteria or fungi within viable tissue implies a serious risk of sepsis and further tissue invasion. Bacterial invasion of viable tissue is readily apparent by histologic study of appropriate tissue samples ( Fig. 38.1 ). Invasive fungal infection presents a somewhat different pattern, in that there is often a wavefront of necrosis that accompanies fungal invasion ( Fig. 38.2 ). Thus the presence of fungal hyphae extending to a boundary between necrotic and viable tissue is considered evidence of fungal invasion of viable tissue. On this basis, infections identified within burn wounds are reported as surface colonization, invasion of necrotic tissue, which may be superficial or deep, and invasion of viable tissue. The responsible surgeon is called immediately when invasion of viable tissue is found. Diagnosis of viral infection of the skin is achieved most efficiently by sampling freshly opened vesicular lesions or the bases of recently ruptured vesicles, and molecular testing by polymerase chain reaction (PCR).
This high-magnification micrograph shows gram-negative rods scattered in the stroma and concentrated in and around a vein that contains a fibrin thrombus. The presence of nuclear staining in stromal cells indicates that the connective tissue surrounding the bacteria is intact. Tissue Gram stain.
This micrograph shows fungal hyphae within necrotic tissue extending close to the boundary with intact, viable dermal tissue. Periodic acid–Schiff stain.
Once septicemia occurs, there is a generalized reaction that includes hypotension, tachycardia, increased hyperthermia or hypothermia, and poor perfusion of the intestines and other viscera. Coagulopathy is also an important complication of sepsis. In addition, once bacteria have gained entrance to the general circulation, tissue infection may develop at distant sites, particularly the lungs. One remarkable case demonstrated clearly the route of dissemination of fatal burn wound sepsis. The patient was admitted in a clinically septic condition 2 weeks after suffering a large burn but died in spite of aggressive therapy. At the time of autopsy there were many areas of bacterial proliferation within the burn wound, invasion of viable tissue deep to the burn eschar, and thrombosis of blood vessels invaded by bacteria at the margin of the necrotic zone. Bacteria were especially numerous within the smooth muscle of veins in the dermis. Lesions of ecthyma gangrenosum were not present in this case. Septic emboli with fibrous organization were seen in pulmonary artery branches in all lobes of the lungs. There were pale foci of necrosis throughout the lungs surrounded by hemorrhage filling the alveoli in a broad zone around each necrotic focus ( Fig. 38.3 ). Very little acute inflammatory reaction was seen in or around the many foci of bland necrosis in the lungs. In these necrotic foci, bacterial proliferation was very prominent within the walls of arteries. A direct hematogenous route of spread of infection from the skin to the lungs seemed clear in this case. Bacteria that had colonized the necrotic tissue associated with the burn wound, over time, apparently became more concentrated and extended into viable tissue, causing local necrosis. They became concentrated in the medial walls of veins at the border between necrotic and viable tissue, caused thrombosis of those vessels, and proceeded to extend into the fresh thrombi. Once the infected thrombi broke loose into the bloodstream, they naturally lodged in small pulmonary artery branches in the lungs, where they continued to grow and to become very prominent in the medial walls of the small pulmonary arterial branches.
This photograph is a slice of the upper lobe of the lung taken at autopsy from a patient who showed clinical signs of sepsis when he developed disseminated infection after multiple wound cultures of open wounds yielded multiple antibiotic-resistant Pseudomonas.
Interestingly, the presence of bacteria concentrated within the walls of arteries or veins has been shown to be a characteristic feature of bacteria forming biofilms in tissues. Growth as biofilms is an alternative to the planktonic growth that occurs in culture medium, which provides strong protection against antibiotics and antiseptic agents, and it is most familiar in growth on solid surfaces such as implants, catheters, and endotracheal tubes. The switch to growth as biofilms occurs in response to small molecular signals that are secreted, typically when bacterial density increases, and stimulate synthesis and secretion of large carbohydrate matrices called alginate as well as metabolic changes and production of multiple toxins, further secretion of the signaling molecules, and sometimes a shift to an elongated phenotype. , It is possible, although research in animal models would be necessary to demonstrate it, that growth of pathogens such as P. aeruginosa in the form of biofilms in blood vessels in the skin predisposes them to become concentrated in the walls of pulmonary blood vessels and to secrete toxins that might contribute to the destruction of neutrophils and lung tissues in the sites of infection in the lungs. Many bacterial species are capable of forming biofilms in tissues, one of which is A. baumannii/haemolyticus. The histologic feature of prominent bacterial growth in the walls of blood vessels has been described in cases of Pseudomonas septicemia in immunosuppressed individuals since the 1960s. In many of those cases, there were foci of secondary infection in multiple organs, whereas foci of infection in organs other than the lungs were strikingly absent in our series of burn autopsies.
It should also be noted that airway obstruction is known to predispose to pneumonia acquired through the airways, by preventing normal clearance of bacteria from the airways and by providing a favorable medium for bacterial growth. Multiple foci of airway obstruction are almost always seen at autopsy in burn patients. , The risk of infection is proportional to the extent of severe burns, the time elapsed before initiation of fluid therapy, the presence of metabolic alterations, the development of immunologic deficiency, the concurrence of trauma, the local evolution of wounds, and the age of the patient. Although many serious infections in burn patients are caused by endogenous flora, and many derive from wound infections present at the time of admission, nosocomial infection is a constant hazard. ,
The problem of burn wound sepsis is amenable to therapy. The strategy of excision of the potentially infected burn wound as early as possible, together with judicious administration of effective antibiotics, has greatly reduced the number of deaths caused by infection in burn patients. Organisms resistant to all available antibiotics are seen in burn patients with increasing frequency, and development of new antibiotics has not kept pace. Patients who are referred for therapy more than 1 week after burn injury often have extensive invasive wound infections and sepsis.
Other microorganisms can also cause life-threatening infection in burn patients. Fungal infection is a continuing problem in patients with large burns, despite the development of more effective systemic antifungal agents. Diagnosis of specific fungal infections by histopathology is difficult, as important morphologic features that allow identification in culture do not occur in tissues, and fungi may display unexpected structural features in injured tissues. Pigmented fungal species generally do not show invasive behavior, and saprophytic species can mimic zygomycetes, which are dangerous pathogens in burn wounds. In one recent case of apparent systemic fungal infection, invasive and systemic dissemination of a pseudofungus of the genus Oomyces was demonstrated. Molecular methods (reverse transcription PCR) were used to identify the organism in this case. Viral infections may also complicate burns. We have also experienced cases in which acquisition or reactivation of herpesvirus infections led to major tissue injury. The risks of infection of a victim of a large burn are somewhat similar to those of an immunosuppressed transplant patient.
It seems likely that the increased availability of molecular diagnostic testing of infectious agents may lead to recognition of additional previously unrecognized causes of invasive infection in burn patients and allow more precise and rapid diagnosis of infection. PCR technology has recently become more available. , A genomic approach to the colonization and invasion of burn wounds may clarify the processes of competition between species as well as preserve an indication of the bacteria present previously. Sequencing of whole bacterial genomes has proved valuable in characterizing nosocomial infection, which previously depended on characterizing bacterial DNA by using pulsed-field gel electrophoresis, and has clarified the modes of action of antibiotics. Recently, several laboratories have reported the results of sequencing of bacterial RNA recovered from burn wounds in experimental animals and humans. This can reveal the current metabolic stasis of the bacteria, indicate whether they are growing as biofilms and whether they are producing alginate or specific toxins, and potentially allow identification of expression of macromolecules that confer resistance to specific groups of antibiotics. ,
Coagulopathy
The burn wound has procoagulant effects and may induce coagulation throughout the circulation (disseminated intravascular coagulation [DIC]). , Tissue necrosis can lead to coagulopathy. Generation of thrombin within the circulation leads to generation of fibrin peptides and stimulates acute inflammatory reactions, including increased vascular permeability and upregulation of adhesion molecules on neutrophils and endothelial cells. Generation of fibrin degradation products may interfere with normal thrombosis, and thrombocytopenia can develop. Activation of the kinin system can stimulate further abnormal vascular permeability and hypotension. Consumption of coagulation factors can lead to abnormal bleeding, which can cause extensive tissue injury secondarily. It is important to note that the acute-phase response to burn injury includes increased synthesis of fibrinogen and factor VIII. During the first 3 to 10 days after burn injury, patients often have greater than normal clotting activity. When DIC occurs, coagulation factors are depleted, including antithrombin. , When DIC occurs in the patient’s terminal course, microscopic fibrin thrombi are seen in many organs at the time of autopsy, most commonly in the lungs, skin, kidneys, and gastrointestinal tract. ,
Review of organ systems affected by burns
Integumentary system
The skin is the site of initial injury in burn patients, and many of the events that lead to dysfunction or failure of other organs begin in the skin. Thermal injury rapidly produces irreversible injury and cell death in all tissues in the skin. In many cases, the burn wound excised within 48 hours of injury shows that the entire dermis and all of the hair follicles are necrotic, but much of the subcutaneous adipose tissue remains viable. It appears that the greater insulating capacity of adipose tissue protects it. In some cases, however, necrosis extends deep into the subcutaneous tissue. In extreme cases, the underlying skeletal muscle may become necrotic. Necrosis of skeletal muscle is especially prominent in the case of electrical injury. Frequently a band-like infiltrate of degenerating polymorphonuclear neutrophils is seen in histologic sections in the midst of a totally necrotic dermis. This suggests that the boundary between necrotic and viable tissue may have extended deeper after the initial burn injury and its inflammatory response. There is experimental evidence that burn wounds often evolve from an initial level of necrosis to a deeper level, even from second to third degree, as a result of poor perfusion of the tissue immediately deep to the initial burn injury. This process of vascular stasis deep to the burn is undoubtedly due in part to the rapid loss of intravascular fluid from the damaged capillaries and venules just below the necrotic burn wound. In addition, there is evidence that neutrophils contribute to this process of burn wound extension.
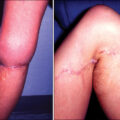
A fortunately rare complication involving the skin of patients with large burns is the development of squamous cell carcinoma during the late phase of recovery. This Marjolin ulcer shows aggressive local invasion but usually can be controlled with adequate local excision.
Respiratory system
Respiratory failure, defined as inability to maintain adequate oxygen saturation while administering 100% oxygen by ventilator, is the immediate cause of death in some burn patients. The causes and mechanisms of respiratory failure are multiple and will be addressed separately, although in many cases more than one mechanism operates. Direct thermal injury to the trachea and bronchi probably does not occur, except in patients exposed to large amounts of steam. In addition to the problems listed later, patients may develop problems related to the airways, such as pneumothorax or interstitial emphysema, aspiration of gastric contents, pulmonary embolism, and nonspecific pulmonary edema caused by increased venous pressure. Lesions of the lungs, which may be multiple, are often found at autopsy in burn patients.
In patients who have clinical evidence of sepsis at the time of death, extensive infection of the lungs is commonly present. Fatal pneumonia is most often seen as a consequence of infection with a highly antibiotic-resistant bacterial strain. Virulent and antibiotic-resistant strains of Pseudomonas or Acinetobacter may produce an angioinvasive infection in the lung, with massive proliferation of bacteria within the walls of pulmonary artery branches and necrosis of nodules of lung tissue in all lobes ( Figs. 38.3–38.5 ). This pattern is similar to that of ecthyma gangrenosum of the skin, which is also characterized by bland necrosis thought to be a result of ischemia. , A similar angioinvasive pattern of pulmonary infection can be seen with generalized infection caused by Aspergillus or similar filamentous fungi.
This micrograph, taken at low magnification, shows a small round pale area of necrotic lung tissue in which no nuclei are staining, surrounded by a zone of congestion and hemorrhage, with very little acute inflammatory reaction. H&E stain.
This high magnification micrograph shows numerous gram-negative rods within the wall of a small pulmonary artery branch and extending into the surrounding pulmonary interstitium. Tissue Gram stain.
Diffuse alveolar damage affects the pulmonary parenchyma in all lobes, although it is often patchy in distribution, and begins with exudation of protein-rich fluid into alveolar spaces. This proteinaceous exudate is a consequence of damage to and increased permeability of both capillary endothelial lining cells and the epithelial type I cells of the alveolar lining. Within hours the exudates form hyaline membranes, which are a histologic hallmark of this disease process ( Fig. 38.6 ). Within a few days the exudate begins to undergo organization by spindle-shaped fibroblasts within alveolar spaces ( Fig. 38.7 ), and collagenous fibrosis develops, which obliterates alveoli and greatly thickens the septa between alveoli and extends between capillaries and the alveolar surface. Macrophages accumulate within alveoli, and alveolar epithelial type II cells multiply. In the late stages there is severe interstitial fibrosis. There are multiple pathogenic mechanisms involved in this process, and it is not yet clear which of these are most significant in patients with burn injury. , The burn injury itself stimulates activation of complement, which could stimulate vascular leakage in the pulmonary bed. These and many other peptides are thought to activate circulating neutrophils, which produce secondary injury to the vascular and epithelial membranes in the lung. , Conversion of xanthine dehydrogenase to xanthine oxidase in the burn wound can cause release of superoxide into the venous circulation, stimulating endothelial injury and oxidative stress in the lung. The neutrophils reacting to the burn wound undergo an oxidative burst, with release of superoxide into the circulation. This process is greatly enhanced if the patient’s course is complicated by ischemic injury to muscle compartments, limbs, or other organs. Lipid peroxidation is a recognized consequence of burn injury. Superoxide can also react with nitric oxide, produced in the wound or the lung, to form peroxynitrite, a highly toxic substance. Thrombin peptides, released during thrombosis of blood vessels in the wound, can also activate neutrophils and stimulate endothelial cells to express adhesion molecules. The kinin system can be activated during thermal injury, with its systemic consequences. When patients develop sepsis, additional pulmonary damage may be produced by release of proinflammatory cytokines and augmentation of the processes that lead to inflammatory injury of the lung. , Neuropeptides, including substance P and calcitonin gene-related peptide, may have a role in increasing vascular leakage in the airways. Finally, the presence of oxygen in high concentration can itself lead to injury manifested as diffuse alveolar damage. Despite this plethora of mechanisms that can lead to pulmonary injury in burn patients, many patients with massive burn injury do not develop clinically apparent respiratory difficulty. The conditions that seem to be most strongly associated with this form of pulmonary injury are delayed fluid resuscitation, limb ischemia, and sepsis.
This section of lung tissue shows bright pink, homogeneous hyaline membranes attached to alveolar walls and septa of alveolar ducts. This is from a 2-year-old patient who died 8 days after a large scald burn. This represents the exudative phase of diffuse alveolar damage, which may be seen in the absence of smoke inhalation injury. H&E stain.
This micrograph shows numerous elongated cells resembling fibroblasts occupying alveolar spaces and secreting connective tissue matrix. This abnormality represents the proliferative phase of diffuse alveolar damage. This patient died 1 month after a large flame burn and had clinical evidence of smoke inhalation injury and acute respiratory distress syndrome. H&E stain.
Smoke inhalation injury commonly complicates burns that occur in burning buildings. The toxic effects of products of combustion directly injure tissues in the lung. These patients are recognized during bronchoscopy by observing hyperemia of the tracheobronchial mucosa and small particles of carbonaceous soot within the airways. Associated findings include facial burns and singed nasal hairs. These patients usually do not require ventilator therapy for several days, but are at high risk of developing respiratory failure, which responds poorly to ventilator therapy and may prove fatal even when the burn injury is small. The mortality of burn injury has been found to be greatly increased when inhalation injury also is present. Experimental studies in sheep and dogs have partially clarified the mechanisms of smoke inhalation injury. In animals, the immediate reactions to inhalation of toxic smoke include detachment of numerous ciliated columnar cells from the tracheobronchial epithelium, massive secretion of mucus by secretory cells, and a more than 10-fold increase in tracheobronchial blood flow. Within a few hours an intense acute inflammatory reaction develops in the airways, with exudation of numerous neutrophils and release of protein-rich fluid that may coagulate within airways, forming occlusive casts ( Fig. 38.8 ). After 48 hours the exudate of neutrophils, which is most intense in the trachea at earlier times, fills many terminal bronchioles and begins to extend into the lung parenchyma ( Fig. 38.9 ). Studies using markers of upper airway mucus, including alcian blue staining and immunostaining for Muc5B, demonstrate the presence of this material in small peripheral airways and alveoli, showing that the obstructive material moves from larger to smaller, more peripheral airways. This inflammatory reaction resolves in the experimental animal, and the epithelium slowly regenerates. However, autopsy evidence suggests that exudation of neutrophils and protein into the airways resolves poorly in humans and may persist for weeks ( Fig. 38.10 ). The loss of airway epithelium, which may be extensive, does not regenerate for long periods. Perhaps because of failure of the mucociliary escalator, mucus can be seen to accumulate around terminal bronchioles focally. Multiple mechanisms may be responsible for the respiratory disease evoked by inhalation of toxic smoke. Factors that may be likely to lead to selective damage to the airways include local release of neuropeptides by afferent C-fibers in the airways and activation of vagal reflexes and activation of proinflammatory processes in reaction to injury to the airway mucosa, particularly local production of interleukin-8. Local production of nitric oxide and other reactive nitrogen species has significant deleterious effects in this form of acute lung injury, according to large animal experiments. , Local activation of thrombin during the formation of fibrin clots and local production of endothelin 1 may further enhance the inflammatory reaction in the airways. , Secretory cells appear to be especially sensitive to smoke inhalation injury. Experimental studies in sheep have demonstrated that activation of poly-adenosyl-ribose polymerase contributes to lung injury after burn and smoke inhalation injury. Obstruction of small bronchi and bronchioles is thought to lead to failure of ventilation of multiple small segments of lung tissue, and inappropriate vasodilation in these poorly ventilated segments may well contribute to the failure of adequate oxygenation. , Treatment with nebulized heparin or tissue plasminogen activator has been found to reduce the degree of airway injury in the ovine model, demonstrating the importance of fibrin polymerization in this model. , Segmental atelectasis and prominent vasodilation in focal areas are features of smoke inhalation injury seen in experimental animals and in patients examined at autopsy after burn injury and smoke inhalation injury. Obstruction of bronchi and bronchioles by mucus, fibrin, and cell debris contributes to respiratory malfunction in experimental animals, and similar obstructive material is seen in human lung tissue at autopsy. , , , , The toxic effects of high concentrations of oxygen may complicate the reaction to injury. A recent review of more than 40 years’ experience at Shriners Children’s Texas found that although clinical evidence of smoke inhalation injury was present in only 14% of patients admitted for recent burns, such evidence was recorded in 66% of the patients who died. Although autopsy cases in which death is caused by smoke inhalation injury without infection have become uncommon, histologic evidence of diffuse alveolar damage is still commonly seen in lung tissue sampled at autopsy.
This micrograph of sheep trachea was sampled 3 hours after experimental smoke inhalation injury by insufflation of cooled cotton smoke under anesthesia. Numerous polymorphonuclear neutrophils appear to be flowing into the tracheal lumen from a gap in the epithelial lining. H&E stain.
This micrograph shows polymorphonuclear neutrophils filling the lumen of a small bronchiole in a sheep 48 hours after experimental smoke inhalation injury. H&E stain.
This micrograph shows a bronchiole in human lung tissue obtained at autopsy 7 days after a large flame burn. The lumen is almost completely obstructed by a mixture of fibrin, mucus, and neutrophils. H&E stain.
Cardiovascular system
Despite the tachycardia and increased output common to patients with burn injury, structural lesions of the heart have been uncommon in our autopsies done on a pediatric population of patients. However, cardiac hypertrophy is a consistent finding at autopsy. Cardiac dilation and clinical evidence of poor myocardial contractility develop in some patients after burn injury. Bacterial endocarditis occurs in occasional patients with sepsis complicating burn injury. Nonbacterial thrombotic endocarditis (marantic endocarditis) has also been seen and may give rise to embolic complications ( Fig. 38.11 ). When the endocardial region of the left ventricle is examined at autopsy, small foci of necrosis associated with local hemorrhage are often observed ( Fig. 38.12 ). Contraction band necrosis is sometimes the only evidence of myocardial injury. These lesions may represent poor perfusion of a tissue with high metabolic demands during terminal episodes of hypotension. In some cases, they may represent the effects of endogenous or exogenous adrenergic agents. Rona and his associates have demonstrated that β-adrenergic agents, at high doses, stimulate the development of small foci of myocyte necrosis and hemorrhage in the subendocardial region of the heart. , This mode of injury is potentially preventable in burn patients.
This micrograph of an H&E-stained cardiac valve shows a deposit of fibrin on the tricuspid valve of a patient who died of sepsis and had nonbacterial thrombotic (marantic) endocarditis diagnosed at autopsy. Gram stains did not reveal any bacteria.
This high-magnification micrograph of heart tissue shows the presence of multiple contraction bands in cardiac myocytes, seen as refractile dark red bands running across muscle fibers. This is an irreversible change that is seen early after ischemic injury causes lethal cell injury, especially when the tissue is reperfused with blood. It can also be seen in the setting of β-adrenergic toxicity. H&E stain.
Urinary system
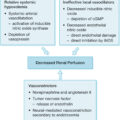
Patients with extensive burn injury, if resuscitated adequately during the first few hours, may have normal renal function throughout their hospital course. It is not uncommon, however, especially when the initial fluid resuscitation was not optimal, or when patients develop episodes of sepsis, for acute renal failure to develop. In such cases, the autopsy frequently reveals evidence of acute tubular necrosis, the morphologic expression of which depends on the timing of the injury. , The morphologic features of acute tubular necrosis include edema of the entire kidney, with a substantial increase in weight, and necrosis of proximal tubular cells with karyolysis, karyorrhexis, and sloughing of the injured cells from the tubular basal lamina. Within 48 hours these events are followed by evidence of regeneration of surviving cells of the renal tubule, which flatten, become basophilic, and undergo mitosis as they migrate to reconstitute the epithelial lining. Clinical renal failure was an independent factor associated with increased mortality in the analysis of prognostic factors in patients with over 80% total body surface area burns in our institution. Patients with renal failure seem to be at especially high risk for the infectious complications of burn injury.
Digestive system and hepatobiliary tract
The association of duodenal ulcers with burn injury, described by Curling, is a classic lesion that still occurs in patients with burn injury, although its incidence is low probably as a result of routine treatment of burn patients with inhibitors of gastric acid secretion. Local mucosal necrosis and hemorrhage, early manifestations of this process, are seen with some frequency. Such defects in the mucosa are often multiple, typically small and round, and can be associated with significant hemorrhage from the exposed blood vessels deep into the lesion. They heal rapidly and rarely lead to serious complications.
The intestinal tract is especially susceptible to ischemic and hypoxic injury, and lesions related to poor perfusion are often found at the time of autopsy. Decreased blood flow in the splanchnic circulation is a well-established physiologic consequence of endotoxemia. , Thus sepsis is associated with an increased risk of intestinal injury. Hypoxic or ischemic injury of the intestinal epithelium, which may be very limited, can lead to translocation of intestinal flora into the mesenteric lymphatic circulation and into the portal venous circulation. Additional factors favoring the escape of bacteria from the intestine include immunosuppression and alterations in the bacterial ecology of the gut. Thus hypotension and hypoxia can also be causes of sepsis. In our autopsy experience, foci of tissue infection in the intestinal tract were very uncommon. The intestinal lesion most commonly seen at autopsy is transverse streaks of hemorrhage in the small intestine in a ladder pattern, associated with focal necrosis of folds of mucosa. This is called superficial hemorrhagic necrosis ( Fig. 38.13 ). Perhaps surprisingly, perforation of the intestinal tract is an uncommon occurrence in patients with burn injury. Occasionally, patients develop pseudomembranous colitis, typically a consequence of infection by toxin-producing Clostridioides difficile ( Fig. 38.14 ). This complication can be minimized by judicious use of antibiotics and screening for the toxin in stool when pseudomembranous colitis is suspected.