Systemic Autoinflammatory Diseases: Introduction
|
Overview of Autoinflammatory Diseases
This chapter focuses on the clinical and immunologic description of the currently known monogenic autoinflammatory syndromes. Most of these syndromes present with predominantly neutrophilic skin eruptions and either fever or systemic inflammation, with elevation of acute-phase reactants during the attacks. Disease-specific organ inflammation often involves the skin, serosal surfaces, the eyes, the inner ear the meninges, the bones, the gastrointestinal tract, lymphadenopathy and more rarely, the vasculature. The characteristic clinical pattern of organ-specific inflammation in the various syndromes can, in most cases, be used to make a clinical diagnosis, which is then confirmed by genetic testing. The syndromes are summarized in this chapter.
 Historic Discovery of Monogenic Autoinflammatory Diseases
Historic Discovery of Monogenic Autoinflammatory Diseases
 Autoinflammatory diseases are clinical disorders marked by episodic flares of abnormally increased systemic and organ-specific inflammation, mediated predominantly by the cells and molecules of the innate immune system, with a significant host predisposition.1 Historically, the term autoinflammatory diseases was first applied to two periodic fever syndromes that are caused by two single gene mutations encoding molecules involved in innate immune pathways. Both disorders lack the presence of antibodies or antigen-specific T cells, which are suggestive of an adaptive immune disorder such as an autoimmune disease. Mutations in MEFV1 encoding the protein, pyrin, cause familial Mediterranean fever2,3 and mutation in the p55 TNF receptor, TNFRSF1, cause the disease previously called familial Hibernian Fever, which prompted a disease name reflecting the molecular defect: TNF receptor-associated periodic syndrome (TRAPS).4 As both disorders also present as recurrent periodic syndromes, the term hereditary (periodic) fever syndromes was used synonymously with autoinflammatory syndromes. However, the genetic discovery of single gene mutations in innate immune regulatory genes that cause episodic inflammation in disorders that do not present with fevers has led to the preference of the more inclusive term of “autoinflammatory diseases.”
Autoinflammatory diseases are clinical disorders marked by episodic flares of abnormally increased systemic and organ-specific inflammation, mediated predominantly by the cells and molecules of the innate immune system, with a significant host predisposition.1 Historically, the term autoinflammatory diseases was first applied to two periodic fever syndromes that are caused by two single gene mutations encoding molecules involved in innate immune pathways. Both disorders lack the presence of antibodies or antigen-specific T cells, which are suggestive of an adaptive immune disorder such as an autoimmune disease. Mutations in MEFV1 encoding the protein, pyrin, cause familial Mediterranean fever2,3 and mutation in the p55 TNF receptor, TNFRSF1, cause the disease previously called familial Hibernian Fever, which prompted a disease name reflecting the molecular defect: TNF receptor-associated periodic syndrome (TRAPS).4 As both disorders also present as recurrent periodic syndromes, the term hereditary (periodic) fever syndromes was used synonymously with autoinflammatory syndromes. However, the genetic discovery of single gene mutations in innate immune regulatory genes that cause episodic inflammation in disorders that do not present with fevers has led to the preference of the more inclusive term of “autoinflammatory diseases.”
 Over the last decade the number of autoinflammatory disorders increased further and the currently known monogenic autoinflammatory disorders are listed in Table 134-1.
Over the last decade the number of autoinflammatory disorders increased further and the currently known monogenic autoinflammatory disorders are listed in Table 134-1.
Inflammatory Pathogenesis | Disease | Gene | Protein | Inheritance Pattern | Disease Onset | Flare/fever Pattern | Specific Organ Inflammation | Treatment |
|---|---|---|---|---|---|---|---|---|
IL-1-mediated | CAPS: | |||||||
FCAS | CIAS1 (1q44) | Cryopyrin | Autosomal dominant | First 6 months of life, cold induced | <24 hours | Skin, eyes, joints | IL-1 blockade | |
MWS | CIAS1 (1q44) | Cryopyrin | Autosomal dominant | Infancy to adolescence | 24–48 hours | Skin, eyes, joints, inner ears, meninges (mild) | IL-1 blockade | |
NOMID | CIAS1 (1q44) | Cryopyrin | Autosomal dominant/de novo | Neonatal or early infancy | Continuous with flares | Skin, eyes, joints, inner ears, meninges, bony epiphyseal hyperplasia | IL-1 blockade | |
DIRA | IL1RN (2q14) | IL-1 receptor antagonist | Autosomal recessive | Neonatal or early infancy | Continuous with flares | Skin, bones, lungs (rare), vasculitis (rare) | Anakinra | |
Partially IL-1-mediated | FMF | MEFV (16p13) | Pyrin | Autosomal recessive | 80% of the cases occur before the age of 20 | 1–3 days | Skin, joints, peritoneum, pleura | Colchicine, rarely IL-1 and TNF blockade or thalidomide if colchicine-resistant |
HIDS | MVK (12q24) | Mevalonate kinase | Autosomal recessive | Median age at onset 6 months | 3–7 days | Skin, eyes, joints, prominent lymph nodes | NSAIDS, corticosteroids, TNF and IL-1 blockade | |
Mevalonic aciduria | MVK (12q24) | Mevalonate kinase | Autosomal recessive | Neonatal or early infancy | Continuous with flares | Lymph nodes, hepatosplenomegaly, eyes microcephaly, cerebellum, GI tract | TNF and IL-1 blockade? Allogeneic bone marrow transplant | |
PAPA | CD2BP1 (15q24) | PSTPIP1 | Autosomal dominant | Early childhood | Common | Skin, joints | Local and systemic corticosteroids, TNF or IL-1 blockade | |
Other pathways/effector pathway not yet known | TRAPS | TNFRSF1A (12p13) | TNF receptor 1 | Autosomal dominant | Median age at onset 3 years | 1–4 weeks | Skin, eyes, joints, peritoneum, pleura | TNF blockade, steroids, IL-1 blockade, colchicine is ineffective |
PGAa | NOD2 (16q12) | Nod2 | Autosomal dominant/de novo | Early childhood | Uncommon | Skin, eyes, joints | NSAIDS, corticosteroids, methotrexate, cyclosporine, TNF or IL-1 blockade | |
Majeed’s syndrome | LPIN2 (18p11) | Lipin-2 | Autosomal recessive | Early infancy (1–19 months) | Weeks–months | Bones, periosteum, anemia | NSAIDS, corticosteroids, interferon-α | |
Early-onset inflammatory bowel disease (IBD) | IL10RA (11q23) IL10RB (21q22) | IL-10 receptor, IL10RB also forms IL-22, -26, -28, -29 receptors | Autosomal recessive | Neonatal or early infancy | Continuous with flares | Colitis with fistula formation, folliculitis in patients with IL10RB mutations | Bone marrow transplantation in severe cases |
 In 1999, the same year of the discovery of the gene causing TRAPS, hyperimmunoglobulin D with periodic fever syndrome (HIDS), another periodic fever syndrome described in the Dutch population was found to be caused by mutations in mevalonate kinase, MVK.5,6 NOD2 mutations initially described in the autosomal dominant disorder Blau syndrome were also found to cause a sporadic form of the disease, early-onset sarcoidosis,7,8 both diseases are now referred to as pediatric granulomatous arthritis (PGA). Pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) is caused by mutations in PSTPIP1.9 The discovery that mutations in NLRP3 (also called CIAS1, NALP3, PYPAF1) cause the spectrum of the cryopyrinopathies, including FCAS, MWS,10 and NOMID,11,12 has not only led to the recognition of a class of novel intracellular “receptor complexes,” the NOD-like receptors (NLRs) that sense intracellular danger, but also gave key insights in the activation and role of IL-1 in coordinating an immune response not only in the cryopyrinopathies but also a wider spectrum of inflammatory conditions in which the genetic cause of the disease has not yet been discovered.13 The discovery that deficiency of IL-1 receptor antagonist (DIRA) is a new neonatal syndrome caused by increased IL-1 signaling due to absence of the inhibitor of IL-1 signaling, the IL-1 receptor antagonist, IL1RN, has further expanded the clinical spectrum of IL-1-mediated organ inflammation.14,15 The pathogenic implications of mutations in LPIN2 causing Majeed’s syndrome,16 an autoinflammatory bone disease,17 are less well understood. Recently, a novel cytokine pathway causing a systemic inflammatory disease in humans was added with the discovery that the absence of signaling of the anti-inflammatory cytokine IL-10, due to a loss of function mutation in the IL-10 receptor, IL10RA or IL10RB, can clinically present with severe early-onset enterocolitis.18
In 1999, the same year of the discovery of the gene causing TRAPS, hyperimmunoglobulin D with periodic fever syndrome (HIDS), another periodic fever syndrome described in the Dutch population was found to be caused by mutations in mevalonate kinase, MVK.5,6 NOD2 mutations initially described in the autosomal dominant disorder Blau syndrome were also found to cause a sporadic form of the disease, early-onset sarcoidosis,7,8 both diseases are now referred to as pediatric granulomatous arthritis (PGA). Pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) is caused by mutations in PSTPIP1.9 The discovery that mutations in NLRP3 (also called CIAS1, NALP3, PYPAF1) cause the spectrum of the cryopyrinopathies, including FCAS, MWS,10 and NOMID,11,12 has not only led to the recognition of a class of novel intracellular “receptor complexes,” the NOD-like receptors (NLRs) that sense intracellular danger, but also gave key insights in the activation and role of IL-1 in coordinating an immune response not only in the cryopyrinopathies but also a wider spectrum of inflammatory conditions in which the genetic cause of the disease has not yet been discovered.13 The discovery that deficiency of IL-1 receptor antagonist (DIRA) is a new neonatal syndrome caused by increased IL-1 signaling due to absence of the inhibitor of IL-1 signaling, the IL-1 receptor antagonist, IL1RN, has further expanded the clinical spectrum of IL-1-mediated organ inflammation.14,15 The pathogenic implications of mutations in LPIN2 causing Majeed’s syndrome,16 an autoinflammatory bone disease,17 are less well understood. Recently, a novel cytokine pathway causing a systemic inflammatory disease in humans was added with the discovery that the absence of signaling of the anti-inflammatory cytokine IL-10, due to a loss of function mutation in the IL-10 receptor, IL10RA or IL10RB, can clinically present with severe early-onset enterocolitis.18
 The gene discovery led to an understanding of the pathogenesis of a wider spectrum of inflammatory conditions. Inflammation in its broadest sense is the response of living organisms to exogenous and endogenous “danger” that evolved to protect organisms from harmful environmental influences with the goal to maintain homeostasis.19 The molecular mechanisms of how organisms “sense” danger are remarkably conserved during evolution and involve receptors that sense harmful exogenous triggers, for example, bacterial cell walls, or viral RNA and DNA that are called pathogen-associated molecular patterns (PAMPs), and a growing number of endogenous triggers released from, for example, dying and stressed tissues that are referred to as damage-associated molecular patterns (DAMPs).19 These molecules get recognized and signal through extracellular (TLRs) and intracellular (NLRs) receptors.20,21
The gene discovery led to an understanding of the pathogenesis of a wider spectrum of inflammatory conditions. Inflammation in its broadest sense is the response of living organisms to exogenous and endogenous “danger” that evolved to protect organisms from harmful environmental influences with the goal to maintain homeostasis.19 The molecular mechanisms of how organisms “sense” danger are remarkably conserved during evolution and involve receptors that sense harmful exogenous triggers, for example, bacterial cell walls, or viral RNA and DNA that are called pathogen-associated molecular patterns (PAMPs), and a growing number of endogenous triggers released from, for example, dying and stressed tissues that are referred to as damage-associated molecular patterns (DAMPs).19 These molecules get recognized and signal through extracellular (TLRs) and intracellular (NLRs) receptors.20,21
 CAPS and PGA are both caused by mutations in the regulatory domain of two intracellular NLR sensors, (1) NLRP3 and (2) NOD2, respectively, the mutations leading to increased sensitivity to danger triggers and increased “danger signaling.” NLRs also form multimolecular complexes that activate immune pathways. In the case of NLRP3 the complex can activate caspase-1, an enzyme that cleaves the inactive proinflammatory cytokines IL-1 and IL-18 into their active form. The IL-1 activating complexes are called inflammasomes, and have provided a molecular link from danger recognition to the activation of IL-1 as a pivotal proinflammatory “alarm” cytokine.
CAPS and PGA are both caused by mutations in the regulatory domain of two intracellular NLR sensors, (1) NLRP3 and (2) NOD2, respectively, the mutations leading to increased sensitivity to danger triggers and increased “danger signaling.” NLRs also form multimolecular complexes that activate immune pathways. In the case of NLRP3 the complex can activate caspase-1, an enzyme that cleaves the inactive proinflammatory cytokines IL-1 and IL-18 into their active form. The IL-1 activating complexes are called inflammasomes, and have provided a molecular link from danger recognition to the activation of IL-1 as a pivotal proinflammatory “alarm” cytokine.
 The inflammasome can be activated by a number of triggers including uric acid crystals, which led to the exploration of the role of IL-1 in gout and a growing number of inflammatory conditions that currently have no known genetic cause.22 Proof of concept studies using IL-1 blockade to treat human disease have confirmed a role for IL-1 in causing gout,23–25 in glucose control in type 2 diabetes26 and a case of Behçet’s disease.27 The role of IL-1 has also been demonstrated in systemic-onset Still’s disease28 and more recently PFAPA, another periodic fever syndrome presenting with aphthous stomatitis, pharyngitis, and cervical adenopathy.
The inflammasome can be activated by a number of triggers including uric acid crystals, which led to the exploration of the role of IL-1 in gout and a growing number of inflammatory conditions that currently have no known genetic cause.22 Proof of concept studies using IL-1 blockade to treat human disease have confirmed a role for IL-1 in causing gout,23–25 in glucose control in type 2 diabetes26 and a case of Behçet’s disease.27 The role of IL-1 has also been demonstrated in systemic-onset Still’s disease28 and more recently PFAPA, another periodic fever syndrome presenting with aphthous stomatitis, pharyngitis, and cervical adenopathy.
Cryopyrinopathies (FCAS, MWS, NOMID)
|
The spectrum of cryopyrinopathies includes (1) the familial cold autoinflammatory syndrome (FCAS; OMIM #120100),29 (2) Muckle–Wells syndrome (MWS; OMIM #191900),30 and (3) the noninherited sporadic disease neonatal-onset multisystem inflammatory disease (NOMID) also called chronic infantile neurologic, cutaneous and arthritis (CINCA) syndrome (OMIM #607115).31 These three clinical disorders were initially described as distinct disorders, but the discovery that the familial disorders FCAS and MWS and the sporadic disease NOMID/CINCA are all caused by autosomal dominant gain of function mutations in NLRP3, which is also called NALP3, CIAS1 or PYPAF110–12 led to the recognition that these disorders form a disease spectrum with FCAS on the mild and NOMID on the most severe end of the spectrum.
The prevalence of CAPS is estimated to be between 1 and 2 patients per 1,000,000 people in Europe and the United States based on known cases in centers studying these patients, but actual epidemiologic studies to assess incidence and prevalence have thus far not been conducted.
NLRP3/CIAS1 encodes the protein, cryopyrin, which belongs to a family of intracellular sensor molecules, the NLRs (NOD-like receptors). Cryopyrin can associate with adaptor proteins to form a multimolecular complex called the NLRP3 inflammasome that activates caspase-1, an enzyme that cleaves pro-IL-1β into its 17-kDa active IL-1 fragment.32 Mutations in cryopyrin result in an “overactive” inflammasome that leads to higher production of active IL-1.12,33–35 The inflammasome can be activated by a number of different triggers including bacterial triggers and molecules released during cell injury or stress [such as adenosine triphosphate (ATP) and uric acid], which leads to formation and activation of the NLRP3 inflammasome and activation of caspase-1, an enzyme that cleaves the proinflammatory cytokine IL-1β into its active form.
 Current Hypothesis of Mechanism
Current Hypothesis of Mechanism A current hypothesis is that wild-type cryopyrin is autoinhibited by a putative conformational interaction between the LRR domain and the NACHT domain. The majority of CIAS1 mutations are thought to disrupt the inactive closed ring conformation of wild-type cryopyrin and facilitate oligomerization of cryopyrin.36 However, about 50% of patients with clinical NOMID/CINCA and a much smaller percentage of FCAS and MWS patients do not have germ-line mutations in NLRP3 in DNA isolated from peripheral blood cells; they are, however, clinically indistinguishable from the NLRP3 mutation-positive patients and therefore a diagnosis of a CAPS should not be excluded on the basis of negative genetic testing. Somatic mosaicism of NLRP3 or the involvement of other genes in the disease pathogenesis has been suggested to account for at least some mutation-negative patients.37
A current hypothesis is that wild-type cryopyrin is autoinhibited by a putative conformational interaction between the LRR domain and the NACHT domain. The majority of CIAS1 mutations are thought to disrupt the inactive closed ring conformation of wild-type cryopyrin and facilitate oligomerization of cryopyrin.36 However, about 50% of patients with clinical NOMID/CINCA and a much smaller percentage of FCAS and MWS patients do not have germ-line mutations in NLRP3 in DNA isolated from peripheral blood cells; they are, however, clinically indistinguishable from the NLRP3 mutation-positive patients and therefore a diagnosis of a CAPS should not be excluded on the basis of negative genetic testing. Somatic mosaicism of NLRP3 or the involvement of other genes in the disease pathogenesis has been suggested to account for at least some mutation-negative patients.37
All CAPS patients present with episodes of fever, urticaria-like eruption, conjunctivitis (Fig. 134-1H), arthralgia, and elevations in acute-phase reactants, but differ in the spectrum of multiorgan disease manifestations and in long-term morbidity and mortality.
Figure 134-1
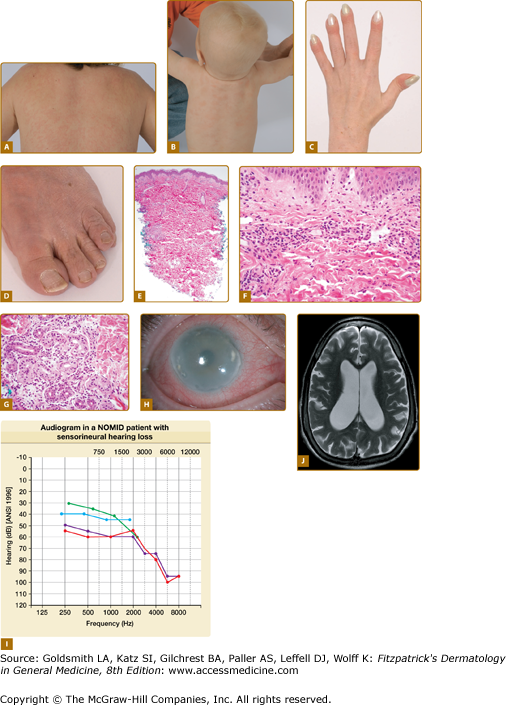
CAPS/NOMID. A. Urticaria-like skin eruption in a patient with NOMID/MWS. B. Urticaria-like skin eruption in an infant with NOMID/MWS. C. Clubbing fingers. D. Wrinkled, excess skin around the toenails and toes. E. At scanning magnification, there is a perivascular and periadnexal inflammatory infiltrate, extending from the superficial dermis to middermis. The epidermal changes are unremarkable. F. The superficial dermal capillaries are dilated and are surrounded by neutrophils. Changes of frank leukocytoclastic vasculitis are absent. G. The eccrine glands are also surrounded and infiltrated by neutrophils. H. Conjunctivitis and corneal clouding due to subcorneal inflammatory infiltrates. I. Sensorineural hearing loss most pronounced at high frequencies. J. Ventriculomegaly and brain atrophy.
In patients with FCAS the episodes are triggered by exposure to cold temperatures or air drafts and present with fevers, eruption, joint pain, conjunctivitis, and headaches. Typical episodes last for 12–48 hours and then resolve. The disease typically starts in early childhood.
Patients with MWS or NOMID typically present with urticaria-like eruptions at birth or within hours of birth; however, in some patients the first symptoms can present later. Patients with MWS have similar symptoms of fevers, urticaria-like eruption, joint pain, conjunctivitis, and headaches; however, in contrast to patients with FCAS, these symptoms can be continuous and are not typically precipitated by cold exposure. Episcleritis and optic disc edema are exceedingly uncommon in FCAS but are seen in some Muckle Wells patients; sensorineural hearing loss typically develops in the second to third decade of life in MWS patients.33 Patients with NOMID/CINCA present with the most severe phenotype. Often their initial presentation is thought to be neonatal sepsis as they present with fevers, urticaria-like eruption, leukocytosis, and high elevation of the acute-phase reactants around birth. Joint swelling and conjunctivitis can develop at birth but usually within the first months of life. CNS inflammation with aseptic leptomeningitis and neutrophilic pleocytosis in the cerebrospinal fluid are common. Severely affected children develop hydrocephalus (Fig. 134-1J), brain atrophy, and cognitive impairment. Patients typically develop sensorineural hearing loss within the first year of life (Fig. 134-1I), and on magnetic resonance imaging (MRI) cochlear enhancement is seen and suggests cochlear inflammation as the cause of hearing loss.35 Conjunctivitis, anterior and posterior uveitis, and subcorneal infiltrates can lead to retinal scarring, corneal clouding, and blindness. Arthralgia is common, true arthritis in most cases is very mild; however, between 50% and 70% of patients with NOMID develop characteristic bony overgrowth of the epiphyses of the long bones.38 Most commonly involved is the epiphyses of the distal femur and proximal tibia of the knee, which frequently leads to joint deformities and contractures. Other features include elevated inflammatory markers, short stature, and leukocytosis.
All CAPS patients present with an urticaria-like skin eruption (Figs. 134-1A and 134-1B). In most cases, the eruption is nonpruritic and is not associated with any particular sensation, but some patients describe the eruption as burning or stinging. The skin eruption is often the first noticeable disease manifestation particularly in the less severe cases and is typically considered to be allergic. The eruption is often present at birth or develops within hours of birth but in FCAS patients can also be discovered later in childhood. The eruption is migratory and nonscarring and can vary in severity from day to day and from patient to patient. A persistent livedoid or cutis marmorata pattern, in addition to the urticarial lesions that come and go, is often seen in older patients with long-standing disease. Other cutaneous manifestations can include soft, doughy palms, soles, fingers and toes (Fig. 134-1D) and clubbing of the finger (Fig. 134-1C) and toenails, without any evidence of pulmonary disease. In contrast to true familial and acquired cold urticaria, the ice cube test is negative in CAPS patients.39,40
The histological features in FCAS, MWS, and NOMID are very similar and are discussed together. Histologically, the epidermis in CAPS is usually unaffected. There may be mild edema of the papillary dermis. The superficial dermal capillaries are dilated. A predominantly perivascular neutrophilic inflammatory infiltrate is present in the superficial dermis and middermis (Fig. 134-1E and 134-1F); however, there is no evidence of vascular destruction or true vasculitis and the number and appearance of mast cells is within normal limits.40–43 The eccrine glands are also surrounded and infiltrated by neutrophils (Fig. 134-1G). The histological finding of neutrophilic urticaria contrasts with the lymphocytic and eosinophilic infiltrate seen in classical urticaria and is an important clue to the diagnosis of CAPS.44
Evidence of systemic inflammation is present with marked leukocytosis, thrombocytosis, and mild anemia of chronic disease. Elevated levels of acute-phase reactants, C-reactive protein, erythrocyte sedimentation rate and serum amyloid A (SAA) levels are typically seen. Eosinophilia can be present and is variable. Careful assessment of central nervous system disease in NOMID/CINCA is indicated. Lumbar punctures are performed in patients with NOMID to diagnose leptomeningitis and to monitor intracranial pressures and leukocytosis, which predominantly consists of neutrophils, and the elevation of protein in the cerebrospinal fluid when patients are on treatment. High-resolution gadolinium-enhanced MRI can show leptomeningeal and cochlear enhancement at various degrees in patients with NOMID. X-rays of the long bones indicate the location of epiphyseal lesions and are used to follow the bony overgrowth in patients with NOMID and MRIs of an individual lesion can be helpful to rule out bone tumors and to distinguish the presence of synovial inflammation which is usually fairly nonimpressive compared to the bony abnormalities in NOMID patients.38
Genetic testing for NLRP3/CIAS1 mutations confirms the diagnosis. Although almost all patients with FCAS and MWS are mutation positive, only 50%–60% of patients with NOMID have germ-line mutations that can be detected on genetic testing; therefore, a negative genetic test does not exclude the diagnosis of CAPS.
In the neonate the differential diagnosis includes benign newborn eruptions, NOMID patients can present more dramatically, mimicking neonatal sepsis or congenital “TORCH” infections. Still’s disease, systemic-onset juvenile idiopathic arthritis (SoJIA), and adult-onset Still’s disease (AOSD) need to be considered in the differential diagnosis; although they typically do not present at birth. Patients with FCAS are often diagnosed with allergies or acquired cold urticaria. The bony lesions in NOMID patients can be painful and can initially resemble other benign or malignant bone tumors.
As treatment with IL-1 blocking agents has radically changed the outcome of these disorders, the prognosis in untreated and treated patients with CAPS differs significantly. In addition, the prognosis of the spectrum of CAPS depends on the disease phenotype. Untreated patients with FCAS usually have a good prognosis without hearing loss and the development of amyloidosis is rare. However, in a few families with cold-induced urticaria, the development of hearing loss and amyloidosis have been reported.45,46 The most severe complications of untreated MWS are hearing loss and the development of amyloidosis in up to 30% in European cohorts. Untreated NOMID/CINCA patients present with early-onset hearing loss usually in the first decade of life, develop mental retardation, progressive vision loss due to progressive optic nerve atrophy, hydrocephalus, cerebral atrophy, and children with severe bony overgrowth and the development of joint contractures can develop limb length discrepancies and severe physical disabilities.47
Despite clinical heterogeneity, all patients with FCAS, MWS, and NOMID respond dramatically and invariably to IL-1 blockade. Studies have shown efficacy of anakinra (Kineret®, recombinant IL-1 receptor antagonist) and the long-acting drugs rilonacept (Arcalyst®, IL-1 Trap, a fusion protein of the IL-1 receptor and the Fc portion of IgG) and canakinumab (Ilaris®, IL-1β-blocking antibody) for the treatment of CAPS. All patients show rapid improvement in clinical and laboratory parameters with treatment.58–63
The drugs are generally well tolerated and these medications have now become the standard of care in the treatment of these conditions. Although anakinra has been used longer in the treatment of CAPS, rilonacept and canakinumab have undergone a development program for orphan diseases and are now FDA approved for the treatment of FCAS and MWS.48,49,64 Consistent with the clinical observations of the varied disease severity in FCAS, MWS, and NOMID/CINCA, increasing doses of IL-1-blocking agents such as anakinra are necessary to control more severe disease. Typically, subcutaneous injections of anakinra at 0.5–1.5 mg/kg/day is sufficient to suppress disease activity in FCAS patients. However, in NOMID patients doses up to 6 mg/kg/day or even 10 mg/kg/day50 have been administered to control disease.
The resolution of symptoms and the achievement of inflammatory remission not only in the blood but also in the specific organs with treatment by IL-1 inhibitors are an important proof of concept that the systemic and organ-specific inflammation seen in these disorders is dependent on IL-1β. Notably, only bony overgrowth changes progress on treatment with IL-1-blocking agents which suggests that the continuation of lesional bone growth is likely independent of IL-1.65
 Deficiency of the IL-1 Receptor Antagonist
Deficiency of the IL-1 Receptor Antagonist
|
 Epidemiology and Etiology
Epidemiology and Etiology DIRA (OMIM #612852) is an autosomal recessive disorder caused by loss of function mutations of the IL-1 receptor antagonist (IL1RN).14,15 The mutations in IL1RN are founder mutations that have so far been described in Newfoundland, the Netherlands, the Middle East (Lebanon), and the northern part of Puerto Rico around Arecibo. The allele frequency for the mutation could be estimated in the founder populations in Newfoundland and Northern Puerto Rico, with the allele frequencies being 0.2% in Newfoundland and 1.3% in Northern Puerto Rico.14 Therefore, in some regions of Puerto Rico the incidence of DIRA may be as high as 1 in 6,300 births.
DIRA (OMIM #612852) is an autosomal recessive disorder caused by loss of function mutations of the IL-1 receptor antagonist (IL1RN).14,15 The mutations in IL1RN are founder mutations that have so far been described in Newfoundland, the Netherlands, the Middle East (Lebanon), and the northern part of Puerto Rico around Arecibo. The allele frequency for the mutation could be estimated in the founder populations in Newfoundland and Northern Puerto Rico, with the allele frequencies being 0.2% in Newfoundland and 1.3% in Northern Puerto Rico.14 Therefore, in some regions of Puerto Rico the incidence of DIRA may be as high as 1 in 6,300 births.
 Pathogenesis
Pathogenesis IL-1 receptor antagonist (IL-1Ra) competes with the proinflammatory cytokines IL-1α and IL-1β for binding sites on the IL-1 type I receptor. While IL-1α and IL-1β lead to the formation of a functional signaling complex and transmission of a signal, IL-1Ra prevents the formation of a signaling complex and acts as a negative regulator blocking IL-1 signaling. The homozygous 3′-truncation mutants of IL1RN and the genomic deletion, including the IL1RN locus seen in patients with DIRA, lead to the production of a nonfunctional IL-1 receptor antagonist protein or the absence of the protein, respectively.
IL-1 receptor antagonist (IL-1Ra) competes with the proinflammatory cytokines IL-1α and IL-1β for binding sites on the IL-1 type I receptor. While IL-1α and IL-1β lead to the formation of a functional signaling complex and transmission of a signal, IL-1Ra prevents the formation of a signaling complex and acts as a negative regulator blocking IL-1 signaling. The homozygous 3′-truncation mutants of IL1RN and the genomic deletion, including the IL1RN locus seen in patients with DIRA, lead to the production of a nonfunctional IL-1 receptor antagonist protein or the absence of the protein, respectively.
 Stimulation of patients’ cells with IL-1 leads to increased production of a number of proinflammatory cytokines and chemokines including IL-6, IL-8, MIP-1-α, and TNF suggesting that IL-1 signaling cannot be blocked in these patients. The patients’ parents and healthy siblings who are heterozygous for the mutation express about half the amount of IL-1Ra compared to healthy controls but do not seem to have a clinical phenotype.
Stimulation of patients’ cells with IL-1 leads to increased production of a number of proinflammatory cytokines and chemokines including IL-6, IL-8, MIP-1-α, and TNF suggesting that IL-1 signaling cannot be blocked in these patients. The patients’ parents and healthy siblings who are heterozygous for the mutation express about half the amount of IL-1Ra compared to healthy controls but do not seem to have a clinical phenotype.
 Clinical Findings
Clinical Findings All currently reported patients with DIRA (n = 10) presented with disease at birth or within 2.5 weeks of age. Fetal distress was present in six of ten reported patients. Pustular eruption, joint swelling, oral mucosal lesions, and pain with movement were common initial disease manifestations. The major clinical findings include pustular skin lesions (eFigs. 134-1.1A and 134-1.1B) and characteristic bone manifestations. Characteristic radiographic findings include balloon-like widening of the anterior rib ends (in all ten patients), periosteal elevation of multiple long bones, and multifocal osteolytic lesions. Less common are heterotopic ossification of the proximal femurs (eFig. 134-1.1C), widening of the clavicles (in two patients), metaphyseal erosions of the long bones (in two patients), and multiple osteolytic skull lesions. Cervical vertebral fusion secondary to collapsing vertebral osteolytic lesions as well as C1, C2 instability due to odontoid nonfusion requiring surgical stabilization were seen. One patient who had developed seizures had a magnetic resonance imaging (MRI) lesion suggesting CNS vasculitis and one patient presented with multiple episodes of thrombosis following placement of a central line. One patient developed bony overgrowth of the epiphyses of the long bones including distal and proximal tibia and femurs distal and proximal radius and humerus reminiscent of lesions seen in NOMID, these lesions remained unchanged on anakinra therapy.
All currently reported patients with DIRA (n = 10) presented with disease at birth or within 2.5 weeks of age. Fetal distress was present in six of ten reported patients. Pustular eruption, joint swelling, oral mucosal lesions, and pain with movement were common initial disease manifestations. The major clinical findings include pustular skin lesions (eFigs. 134-1.1A and 134-1.1B) and characteristic bone manifestations. Characteristic radiographic findings include balloon-like widening of the anterior rib ends (in all ten patients), periosteal elevation of multiple long bones, and multifocal osteolytic lesions. Less common are heterotopic ossification of the proximal femurs (eFig. 134-1.1C), widening of the clavicles (in two patients), metaphyseal erosions of the long bones (in two patients), and multiple osteolytic skull lesions. Cervical vertebral fusion secondary to collapsing vertebral osteolytic lesions as well as C1, C2 instability due to odontoid nonfusion requiring surgical stabilization were seen. One patient who had developed seizures had a magnetic resonance imaging (MRI) lesion suggesting CNS vasculitis and one patient presented with multiple episodes of thrombosis following placement of a central line. One patient developed bony overgrowth of the epiphyses of the long bones including distal and proximal tibia and femurs distal and proximal radius and humerus reminiscent of lesions seen in NOMID, these lesions remained unchanged on anakinra therapy.
eFigure 134-1.1
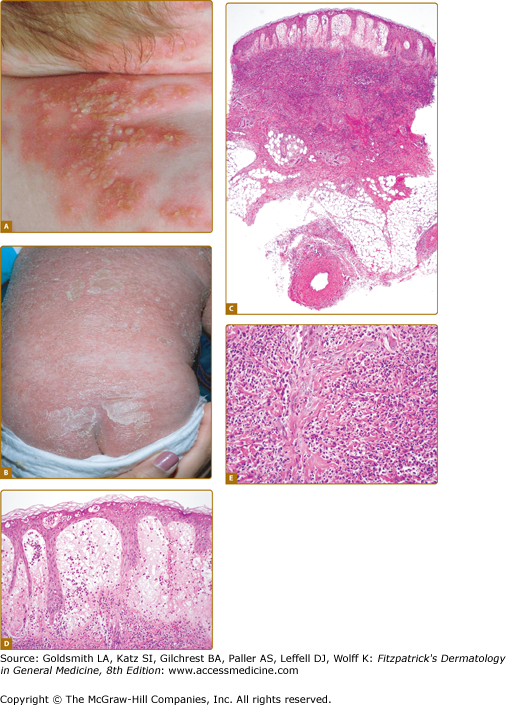
DIRA. A. Grouping of small pustules over erythematous patches on the skin of a child with DIRA. (From Aksentijevich I et al: An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Eng J Med 360:2426-2437, 2009, with permission. © 2009 by the NEJM.) B. Generalized pustulosis, diffuse erythema, and crusting on the skin of a child with DIRA. (Used with permission from Joost Frenkel.) C. At scanning magnification, there is marked edema of the papillary dermis and subepidermal vesiculation. An intense inflammatory infiltrate is present throughout the entire thickness of the reticular dermis. D. The epidermal changes are characterized by the presence of intraepidermal neutrophils and neutrophilic pustules. There is prominent edema of the papillary dermis and a dense neutrophilic infiltrate in the reticular dermis. E. An intense neutrophilic inflammatory infiltrate is present throughout the dermis.
 Cutaneous Lesions
Cutaneous Lesions The severity of the skin findings can vary widely. Cutaneous pustulosis can range from normal skin with rare individual pustules over discrete crops of pustules (eFig. 134-1.1A) to generalized severe pustulosis or ichthyosiform lesions (eFig. 134-1.1B). Oral mucosal vesicular or aphthous ulcers early in life were seen in some patients but do not seem to recur later in life. Nail changes including onychomadesis and pits were also seen in four patients.
The severity of the skin findings can vary widely. Cutaneous pustulosis can range from normal skin with rare individual pustules over discrete crops of pustules (eFig. 134-1.1A) to generalized severe pustulosis or ichthyosiform lesions (eFig. 134-1.1B). Oral mucosal vesicular or aphthous ulcers early in life were seen in some patients but do not seem to recur later in life. Nail changes including onychomadesis and pits were also seen in four patients.
 Dermatopathology
Dermatopathology Histologically, the epidermal changes in DIRA are characterized by the presence of intraepidermal neutrophils and neutrophilic pustules (eFig. 134-1.1D). Marked papillary dermal edema is commonly present and may be associated with subepidermal vesiculation (eFig. 134-1.1E). An intense neutrophilic inflammatory infiltrate is present throughout the dermis and may extend to involve the superficial subcutis (eFig. 134-1.1F). Changes of frank vasculitis are absent in most patients; however, vasculitis was observed in the connective and fat tissue adjacent to bone in one patient.14
Histologically, the epidermal changes in DIRA are characterized by the presence of intraepidermal neutrophils and neutrophilic pustules (eFig. 134-1.1D). Marked papillary dermal edema is commonly present and may be associated with subepidermal vesiculation (eFig. 134-1.1E). An intense neutrophilic inflammatory infiltrate is present throughout the dermis and may extend to involve the superficial subcutis (eFig. 134-1.1F). Changes of frank vasculitis are absent in most patients; however, vasculitis was observed in the connective and fat tissue adjacent to bone in one patient.14
 Laboratory Tests and Special Tests (Including Imaging Studies)
Laboratory Tests and Special Tests (Including Imaging Studies) Inflammatory markers including ESR and CRP in untreated patients are very elevated accompanied by leukocytosis and thrombocytosis. A skeletal survey looking for periosteitis and osteolytic bone lesions should be performed in suspected cases and bone scans may be needed. A skin biopsy can reveal characteristic neutrophilic lesions and genetic testing confirms the diagnosis.
Inflammatory markers including ESR and CRP in untreated patients are very elevated accompanied by leukocytosis and thrombocytosis. A skeletal survey looking for periosteitis and osteolytic bone lesions should be performed in suspected cases and bone scans may be needed. A skin biopsy can reveal characteristic neutrophilic lesions and genetic testing confirms the diagnosis.
 Differential Diagnosis
Differential Diagnosis The differential diagnosis in the perinatal period includes septic osteomyelitis, DIRA can mimic congenital ichthyosiform conditions associated with immunodeficiencies, or resemble mutation-negative NOMID; however, hearing loss, conjunctivitis or other forms of eye inflammation have not yet been observed. One patient developed interstitial lung disease. The bone lesions can resemble the benign, often self-limited cortical hyperostosis.51,52 Based on skin findings, the disease has been mistaken as pustular psoriasis, which typically does not present in newborns.53
The differential diagnosis in the perinatal period includes septic osteomyelitis, DIRA can mimic congenital ichthyosiform conditions associated with immunodeficiencies, or resemble mutation-negative NOMID; however, hearing loss, conjunctivitis or other forms of eye inflammation have not yet been observed. One patient developed interstitial lung disease. The bone lesions can resemble the benign, often self-limited cortical hyperostosis.51,52 Based on skin findings, the disease has been mistaken as pustular psoriasis, which typically does not present in newborns.53
 Complications and Prognosis/Clinical Course
Complications and Prognosis/Clinical Course In untreated patients the mortality is high in the newborn period. If immunosuppressive therapy is not initiated, patients can develop a severe inflammatory response syndrome leading to multiorgan failure and death. Another patient died of cardiopulmonary complications due to chronic lung disease.14
In untreated patients the mortality is high in the newborn period. If immunosuppressive therapy is not initiated, patients can develop a severe inflammatory response syndrome leading to multiorgan failure and death. Another patient died of cardiopulmonary complications due to chronic lung disease.14
 Treatment and Prevention
Treatment and Prevention Treatment with anakinra can be life saving. Anakinra is a recombinant IL-1 receptor antagonist, the recombinant form of the protein these children are missing. In all patients, response to anakinra was immediate, leading to the resolution of clinical symptoms, including eruption and osteolytic lesions and normalization of the acute-phase reactants.14,15 Prenatal diagnosis, within areas where the disease might be common, may be indicated in order to prepare the families and care team for the diagnosis which can prevent costly and invasive workup and lead to timely treatment.
Treatment with anakinra can be life saving. Anakinra is a recombinant IL-1 receptor antagonist, the recombinant form of the protein these children are missing. In all patients, response to anakinra was immediate, leading to the resolution of clinical symptoms, including eruption and osteolytic lesions and normalization of the acute-phase reactants.14,15 Prenatal diagnosis, within areas where the disease might be common, may be indicated in order to prepare the families and care team for the diagnosis which can prevent costly and invasive workup and lead to timely treatment.
Familial Mediterranean Fever
|
Familial Mediterranean fever (FMF; OMIM #249100) is an autosomal recessive disease caused by mutations in MEFV (MEditerranean FeVer), the gene encoding the protein pyrin.2,3 FMF is the most common monogenic autoinflammatory disease. FMF is most common among populations around the Mediterranean Sea. Clinical symptoms of FMF present in most patients before the age of 20 and therefore the diagnosis is mainly made by pediatricians.
 Molecular Genetics
Molecular Genetics More than 70 mutations have been described in FMF but five founder mutations: (1) E148Q in exon 2 and (2) M680I, (3) M694V, (4) M694I, and (5) V726A in exon 10, account for 70%–80% of cases occurring in patients of Mediterranean ancestry.54 The prevalence of clinical cases with FMF varies widely but is most frequent in non-Ashkenazi Jews (~1:248 to 1:1,000),55 Armenians (~1:500), and populations of Arab (~1:2,600) and Turkish ancestry (~1:1,000).56 The carrier rate for the four most common mutations in the Jewish populations is estimated to be between 0.06 and 0.22,57 and in the Turkish population is 0.2.58,59 The high frequency of the carrier rate in some populations has led to suspicions of a selective advantage of the carrier state to a broad class of bacterial pathogens.60 Although at a much lower prevalence, FMF has been described in populations different from the classically affected ones such as Anglo-Saxons and Germans,61 Afghans,62 Indians and Chinese,61 Italians,63–65 Spanish,66 French, Greeks64,67 and Cypriots.68 The male–female ratio is about 1.5 to 2.0:1 and the higher prevalence in males is thought to be partially due to less access to medical care and a higher rate of misdiagnosis in women in some countries with the highest disease prevalence.69,70 Although single heterozygous mutation in MEFV do not typically cause disease in most patients, recent reports suggest that a number of patients with clinically typical FMF have only one identifiable mutation in MEFV and should be treated appropriately.71,72
More than 70 mutations have been described in FMF but five founder mutations: (1) E148Q in exon 2 and (2) M680I, (3) M694V, (4) M694I, and (5) V726A in exon 10, account for 70%–80% of cases occurring in patients of Mediterranean ancestry.54 The prevalence of clinical cases with FMF varies widely but is most frequent in non-Ashkenazi Jews (~1:248 to 1:1,000),55 Armenians (~1:500), and populations of Arab (~1:2,600) and Turkish ancestry (~1:1,000).56 The carrier rate for the four most common mutations in the Jewish populations is estimated to be between 0.06 and 0.22,57 and in the Turkish population is 0.2.58,59 The high frequency of the carrier rate in some populations has led to suspicions of a selective advantage of the carrier state to a broad class of bacterial pathogens.60 Although at a much lower prevalence, FMF has been described in populations different from the classically affected ones such as Anglo-Saxons and Germans,61 Afghans,62 Indians and Chinese,61 Italians,63–65 Spanish,66 French, Greeks64,67 and Cypriots.68 The male–female ratio is about 1.5 to 2.0:1 and the higher prevalence in males is thought to be partially due to less access to medical care and a higher rate of misdiagnosis in women in some countries with the highest disease prevalence.69,70 Although single heterozygous mutation in MEFV do not typically cause disease in most patients, recent reports suggest that a number of patients with clinically typical FMF have only one identifiable mutation in MEFV and should be treated appropriately.71,72
 Pathogenesis
Pathogenesis MEFV encodes the protein pyrin, which is composed of a PYRIN, B-box, coiled–coiled, and the B30.2 (PRYSPRY) domain. Pyrin also belongs to the human tripartite motif (TRIM) family that comprises 70 members that all share the PRYSPRY domain. The most severe mutations in FMF (M680L, T681I, and M694V/L/I) are located in exon 10 and mapped to conserved Fc-containing residues that form conserved hot spots within the B30.2 (PRYSPRY) domain.73 A similar area on TRIM5α, another member of this family, confers viral restriction to HIV and murine leukemia virus.74,75 Interestingly, the FMF mutations in some cases represent a retro-change to an ancestral haplotype seen in other species which has led to speculations that pyrin may have a role in the recognition of pathogens.76 However, the inflammatory pathways induced by mutated pyrin in FMF are not fully understood. In a murine pyrin-knockout model, increased interleukin (IL)-1 secretion is observed and the transfection of wild-type murine pyrin back into murine monocytes suppresses IL-1β production,60 which led to the hypothesis that wild-type pyrin may be anti-inflammatory. Pyrin may also act as a transcription factor as pyrin has two nuclear localization motifs.2,77 Pyrin can be cleaved by caspase-1 and cleaved pyrin can translocate to the nucleus.78 The N-terminal fragment may activate NF-κB through increased calpain-mediated degradation of IκB-α. Pyrin is also found in the cytoplasm of monocytes where it colocalizes with microtubules79 that may explain why colchicine, which can destabilize microtubules, is so effective in the treatment of FMF.80 Pyrin also interacts with ASC, the apoptosis-associated speck-like protein with a caspase-recruitment domain (CARD)—a molecule involved in the formation of an inflammasome through homotypic interactions with pyrin through its pyrin domain and with NLRP proteins and inflammatory caspases through the CARD domain.21 Pyrin’s binding to ASC may lead to sequestration of ASC or its direct binding to caspase-1 may prevent formation of other functional NLR inflammasome complexes81; in both instances, the interaction would lead to a reduction of IL-1 production. Most of the disease-causing mutations in the MEFV gene have been found in the caspase-binding area of the B30.2/PRYSPRY domain, and lead to decreased pyrin binding to ASC. One hypothesis assumes that when pyrin is mutated, ASC is not sequestered and is available to assemble with cryopyrin to form the NALP3 inflammasome, which then leads to increased secretion of proinflammatory cytokines. Another hypothesis suggests that wild-type pyrin may be proinflammatory by forming an IL-1β-activating platform similar to the NLRP3 inflammasome.82 Mutant pyrin in that setting would have a further increased role of inflammasome activation. In addition, wild-type pyrin that is overexpressed in HeLa cells increases ASC speck formation and, paradoxically, increase the survival of these cells.83
MEFV encodes the protein pyrin, which is composed of a PYRIN, B-box, coiled–coiled, and the B30.2 (PRYSPRY) domain. Pyrin also belongs to the human tripartite motif (TRIM) family that comprises 70 members that all share the PRYSPRY domain. The most severe mutations in FMF (M680L, T681I, and M694V/L/I) are located in exon 10 and mapped to conserved Fc-containing residues that form conserved hot spots within the B30.2 (PRYSPRY) domain.73 A similar area on TRIM5α, another member of this family, confers viral restriction to HIV and murine leukemia virus.74,75 Interestingly, the FMF mutations in some cases represent a retro-change to an ancestral haplotype seen in other species which has led to speculations that pyrin may have a role in the recognition of pathogens.76 However, the inflammatory pathways induced by mutated pyrin in FMF are not fully understood. In a murine pyrin-knockout model, increased interleukin (IL)-1 secretion is observed and the transfection of wild-type murine pyrin back into murine monocytes suppresses IL-1β production,60 which led to the hypothesis that wild-type pyrin may be anti-inflammatory. Pyrin may also act as a transcription factor as pyrin has two nuclear localization motifs.2,77 Pyrin can be cleaved by caspase-1 and cleaved pyrin can translocate to the nucleus.78 The N-terminal fragment may activate NF-κB through increased calpain-mediated degradation of IκB-α. Pyrin is also found in the cytoplasm of monocytes where it colocalizes with microtubules79 that may explain why colchicine, which can destabilize microtubules, is so effective in the treatment of FMF.80 Pyrin also interacts with ASC, the apoptosis-associated speck-like protein with a caspase-recruitment domain (CARD)—a molecule involved in the formation of an inflammasome through homotypic interactions with pyrin through its pyrin domain and with NLRP proteins and inflammatory caspases through the CARD domain.21 Pyrin’s binding to ASC may lead to sequestration of ASC or its direct binding to caspase-1 may prevent formation of other functional NLR inflammasome complexes81; in both instances, the interaction would lead to a reduction of IL-1 production. Most of the disease-causing mutations in the MEFV gene have been found in the caspase-binding area of the B30.2/PRYSPRY domain, and lead to decreased pyrin binding to ASC. One hypothesis assumes that when pyrin is mutated, ASC is not sequestered and is available to assemble with cryopyrin to form the NALP3 inflammasome, which then leads to increased secretion of proinflammatory cytokines. Another hypothesis suggests that wild-type pyrin may be proinflammatory by forming an IL-1β-activating platform similar to the NLRP3 inflammasome.82 Mutant pyrin in that setting would have a further increased role of inflammasome activation. In addition, wild-type pyrin that is overexpressed in HeLa cells increases ASC speck formation and, paradoxically, increase the survival of these cells.83
FMF typically presents with 1–3-day episodes of fever with serositis, which typically consists of sterile peritonitis, but pleuritis, arthritis, and eruption can also be present. The arthritis attacks can last up to 1 week. The frequency of the attacks can vary from once weekly to once every few years. Physical exertion and emotional stress can induce these attacks, but in many instances there are no obvious triggers associated with the onset of attacks. In infants, particularly children under the age of 2, fever alone can be the initially presenting feature which typically progresses to more classic features within 3 years,84 which may be complicated by the development of amyloidosis.22 Abdominal pain is the second most common clinical finding in FMF presenting at some time in over 90% of patients. The peritonitis can be severe and mimic a surgical abdomen with a rigid abdominal wall, rebound tenderness, and absent bowl sounds. Not uncommonly, FMF patients have a history of a nondiagnostic exploratory laparoscopy and/or laparotomy. Small peritoneal effusions can be seen on abdominal CT and likely present an inflammatory exudate. Pleurisy is also common and is usually unilateral. A sharp chest pain, worse on inspiration and coughing, can give a clue to the diagnosis.
Symptomatic pericarditis during the attacks occur but are rare,85,86 but on echocardiogram small, nonclinically significant effusions can be present during attacks.
Other rare clinical features of FMF include, protracted febrile myalgia, which presents with up to 6-week episodes of debilitating muscle pain, fever and elevation of acute-phase reactants but normal CK levels.87 Inflammation of the tunica vaginalis, an embryological remnant of the peritoneal membrane,88 can lead to severe scrotal pain and ischemic testicular necrosis and rarely aseptic meningitis have been seen.89
Erysipeloid erythema is the most characteristic eruption of FMF, but it is rarely present in isolation.90 These lesions appear on the lower limbs, usually below the knee. They tend to affect the dorsum of the foot and ankle and lower extremity. They consist of warm, tender, swollen erythematous plaques, 10–15 cm in diameter, with well-defined borders (Fig. 134-2A). The lesions may be bilateral, may be initiated by prolonged walking, and subside within 24–48 hours. Other cutaneous manifestations described in FMF include a localized edema affecting the upper or lower limbs, face, or neck, and subcutaneous nodules.91 In children, episodic scattered nonspecific purpuric papular lesions that affect the face, trunk, or extremities are more frequently observed.92 A higher incidence of vasculitic purpura, including Henoch–Schönlein purpura, has also been described in FMF patients in some case series.69
Figure 134-2
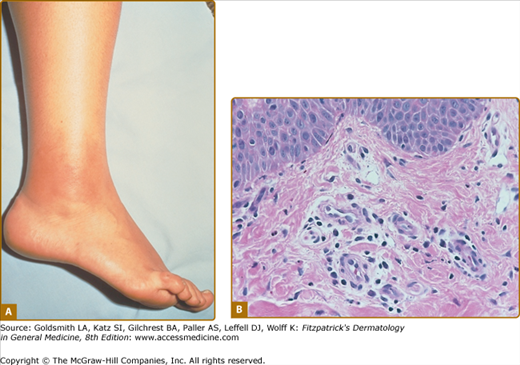
FMF. A. Erysipelas-like eruption on right ankle showing sharply demarcated, swollen, bright erythematous skin. (From Azizi E et al: Cutaneous manifestations of Familial Mediterranean Fever. Arch Dermatol 112:364–366, 1976, with permission. © 1976 by the American Medical Society.) B. Histology of erysipeloid eruption showing a perivascular inflammatory infiltrate in the superficial dermis. (Used with permission from Dan Kastner.)
Biopsies of erysipeloid skin eruptions are rarely reported; on biopsies, a perivascular and interstitial inflammatory infiltrate composed of predominantly neutrophils and lymphocytes in the dermis, with prominent edema of the papillary dermis and dilatation of superficial dermal capillaries, can be seen. The epidermal changes may include mild hyperkeratosis and acanthosis90,91 (Fig. 134-2B). In one series, direct immunofluorescence studies show deposition of complement C3 in the vessel wall of the small superficial dermal vessels in all cases and deposition of fibrinogen and IgM in some case.
During the inflammatory attacks nonspecific markers of inflammation, the acute-phase reactants, ESR, CRP, and SAA levels are typically elevated.93 Mild hypergammaglobulinemia including elevation of IgD, mostly in patients who are homozygous for the M694V mutations, can be seen during as well as in between attacks.94 In between the febrile attacks, elevation of the acute-phase reactants can persist—a finding that has also been observed in heterozygous carriers for the mutation.95,96 Similar to many other autoinflammatory diseases, autoantibodies to ANA, RF, and ANCA are typically not found, which initially led to the hypothesis that these disorders are caused by an innate immune defect.








