Surgery of Neurofibromatosis in the Pediatric Patient
McKay McKinnon
DEFINITION
Patients with cutaneous neurofibromatosis (NF), the most common of the myriad of phenotypic manifestations of the disease (and Von Recklinghausen’s original case reports), can usually expect permanent relief of individual lesions by full-thickness excision. For those patients with so-called plexiform NF and/or malignant peripheral nerve sheath tumors (MPNST), the puzzlement of a permanent treatment (medical or surgical) has remained largely unsolved for most patients.
For well over a century, the textbook recommendation for children with NF1 plexiform tumors has been to defer surgery until after puberty or until serious morbidity is evident. When surgery has been deemed necessary, it has most often been a superficial resection of the tumor, commonly referred to as “debulking.” Subsequent recurrence of tumor has led to a default strategy of withholding surgery for pediatric patients, based upon fear of iatrogenic injury to normal structures, uncontrollable hemorrhage, and perceived inability to achieve permanent tumor removal.
Over the past 30 years, the author has observed that radical resection of plexiform tumors in adults has resulted in permanent “nonrecurrence” of nearly all tumors, even those over 50 kg and some MPNST.1 Despite a higher propensity for active tumor growth in children and adolescents, it seemed logical to extend the concept of radical surgical resection to pediatric patients.
ANATOMY
Craniofacial
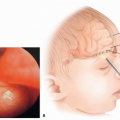
Most facial and frontotemporal scalp tumors derive from sensory branches of cranial nerve V. The ophthalmic division of the trigeminal nerve (FIG 1A) includes some sensory nerves that are within the musculofascial cone (annulus of Zinn) of the orbit. This area presents special risks of injury to the optic nerve and the extraocular muscles and should be avoided.
Orbital NF is often accompanied by a defect of the sphenoid greater wing, which may permit herniation of the temporal lobe into the orbit and subsequent orbital dystopia, pulsatile exophthalmos or enophthalmos, and pressure on the globe and optic nerve. Destruction of bone, ligaments, fascia, muscle, and skin may be present.
Tumor confined to the optic nerve (optic glioma) deserves neurosurgical management.
Motor nerves are not intrinsically involved with NF tumor.
NF tumors of the mandible, parotid, ear, and temporal scalp develop largely from the mandibular division of the trigeminal nerve, including the auriculotemporal nerve.
Tumors of the neck and posterior scalp derive from upper cervical sensory nerves (C1-C4), including the occipital nerves. Lower cervical nerves (C4-C8, sensory and motor) constitute most of the brachial plexus.
Trunk and extremity
Plexiform NF can exist anywhere from the spinal cord to the sensory nerve terminus in the skin, bone, or viscera (FIG 1B).
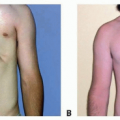
Plexiform tumors most commonly appear between the deep fascia and the skin but may involve and destroy skin, muscles, bones, joints, and visceral organs. Careful dissection proximal to the tumor mass (toward their CNS origin) often reveals the specific sensory nerve origin. Large truncal lesions over time can develop paraspinal hypervascularity, which resembles an arteriovenous malformation. Scoliosis is common with posterior trunk lesions, and limb hyperplasia is common with extremity lesions.
PATHOGENESIS
NF is an autosomal dominant condition, which can be attributed to a defect on chromosome 17, but mosaicism can also occur. The consequential lack of neurofibromin (a tumor inhibitor) permits growth of myelinated axonal tumors in any bodily location. Tumor types are cutaneous or plexiform. So-called plexiform tumors are solid tumors, which have an obligatory angiogenic influence, similar to other solid tumors. They express multiple angiogenic and neurogenic factors. Puberty, pregnancy, and childhood growth spurts can exacerbate their growth. Tumor vascularity increases proportionate to tumor growth. Transformation of benign NF tumors into MPNST can occur in plexiform tumors. This transformation may have a direct correlation to numerical tumor cell replication events.
Tumor vessels in NF are pathologic arteries that reveal absent media and abnormally thin adventitia. Established tumors can exhibit vascular lakes and/or nests of abnormal vessels resembling a congenital vascular malformation.
NATURAL HISTORY
Neurofibromas, once established as plexiform tumors, do not regress spontaneously. Their growth is unpredictable and nonregular, including possible onset throughout a patient’s life of new lesions. Malignant transformation is more common in truncal/extremity lesions but is also unpredictable. Café au lait cutaneous spots do not necessarily signal
plexiform tumors, although they themselves represent microscopic tumor. If present at birth, plexiform tumors may have already inflicted defective tissue development of the newborn.
Eventual destruction by tumor of adjacent skin, bone, and other tissue should be expected without intervention. Hyperplasia and/or fragility of bone occurs in extremity lesions. Craniofacial tumors can also produce osteoclastic or osteoblastic pathology, likely as a consequence of tumor growth rate. Tumors involving the sphenoid and orbit can cause progressive orbital dystopia, blepharoptosis, and blindness. Paraspinal tumors inflict progressive scoliosis on the growing patient.
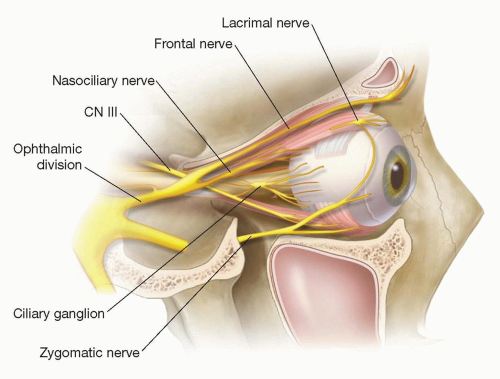 FIG 1 • Nerves of the orbit. |
PATIENT HISTORY AND PHYSICAL FINDINGS
The myriad presentations of NF tumors should not deter accurate diagnosis, even in infancy, by the observant neurologist or experienced surgeon. If doubt of the diagnosis exists, a biopsy should be performed with exception at least of the optic nerve.
Physical findings are too numerous to justify an inclusive list, but palpable tumor, pain, and confirmatory imaging should precede surgical intervention in virtually all cases.
IMAGING
Pediatric patients with suspicion of plexiform NF tumor should have an MRI study, preferably with and without contrast. MRI of the CNS is appropriate as an early screening measure for all NF patients, even those without known tumors. T2- and STIR-weighted images usually give the clearest depiction of NF tumor.
MRI, especially with higher Tesla power, can frequently reveal the specific cranial or spinal nerve involved with tumor. This information (nerve mapping) should be sought out with the neuroradiologist.
High-resolution CT scans should only be ordered when and if the patient with skeletal destruction or high risk of skeletal pathology is being evaluated preoperatively.
DIFFERENTIAL DIAGNOSIS
NF tumors have been mistaken for vascular malformations, particularly by their hypervascular imaging on CT or MRI.
Hyperpigmentation of NF lesions has been confused by the inexperienced physician with congenital nevus. A biopsy can readily produce the correct diagnosis.
Most patients, even infants, with NF will present with at least three or more cutaneous lesions and/or café au lait lesions, axillary freckling, ocular Lisch nodules, or a subcutaneous plexiform mass.
Congenital ptosis can usually be differentiated from NF-derived ptosis by detection of an orbital mass, presence of enophthalmos or exophthalmos, and by MRI findings. Rapid increase of ptosis may be associated with orbital malignancy such as rhabdomyosarcoma, for which immediate incisional or excisional biopsy is required.
Diagnosis of MPNST or other orbital malignancies cannot be reliably made by symptoms, by imaging, or by clinical examination.
NONOPERATIVE MANAGEMENT
This default management decision is historically common but deserves renewed challenge because it is controversial. Observation of growing NF tumors is patently nontherapeutic yet may be justified if surgery is high risk, the patient has significant comorbidity, or the tumor has not reached a level of irreversible morbidity to vital structures. The surgeon and his or her medical colleagues should be prepared to intervene to prevent irreversible morbidity, even in young patients.
SURGICAL MANAGEMENT
An accurate diagnosis is the sine qua non of surgical management. With plexiform NF this should include determination of the tumor nerve origin(s), the degree of vascularity, bony deformity, proximity to other vital anatomy, and degree of skin destruction. In the craniofacial tumors, accurate diagnosis also should determine presence of brain
herniation into the orbit (or elsewhere), orbital dystopia, ptosis vs pseudoptosis, ophthalmic pathology, and airway and carotid/jugular risks.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree