NF2, and schwannomatosis are summarized in Table 24-2. Virtually all of those with NF1 develop NFs, and 50% of children with NF1 have at least one, if not more, NF by the age of 10 years. In children under 10 years of age, NFs often have exclusive plexiform features.
TABLE 24-1. Mesenchymal tumor and tumor-like lesions of the dermis and subcutis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 24-2 Clinical and pathologic features of neurofibromatoses | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
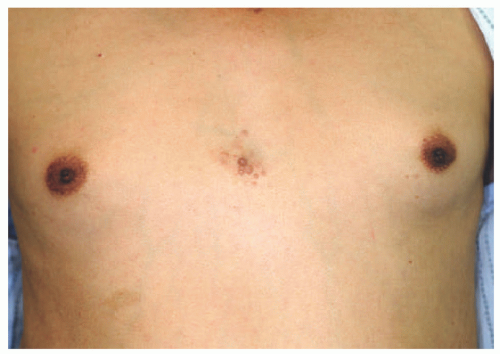 FIGURE 24-1. Neurofibroma. Small pedunculated papules on the trunk. |
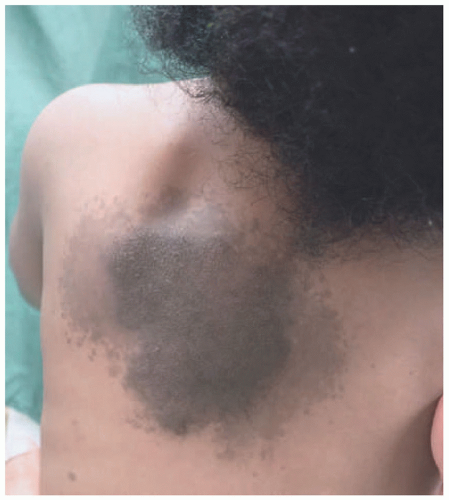 FIGURE 24-2. Plexiform neurofibroma in a patient with neurofibromatosis type 1. |
within the stroma. Nuclear pleomorphism and sclerosis are two uncommon features.18,19 It is uncommon to encounter a solitary NF in an individual less than 20 years of age.
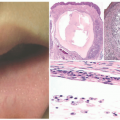
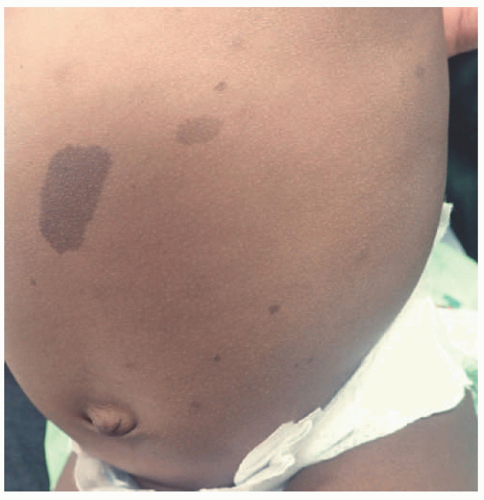 FIGURE 24-3. Large café au lait spots in a patient with neurofibromatosis type 1. |
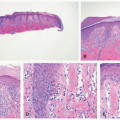
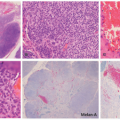
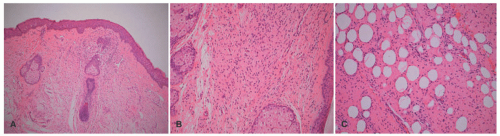 FIGURE 24-4. A-C, Neurofibroma (NF) from the chin of a 12-year-old male with NF1. A, The diffuse NF shows the growth between pilosebaceous units. B, The degree of cellularity can be quite variable from one focus of the same tumor to another. C, Extension into the subcutis demonstrates the so-called honeycomb pattern that is usually made in reference to dermatofibrosarcoma protuberans. |
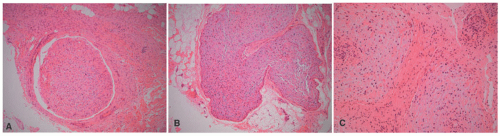 FIGURE 24-5. A-C, Neurofibroma from the scalp of an 11-year-old female with NF1. A, Plexiform profile is surrounded in part by diffuse neurofibroma. B, Serpentine architecture is displayed in these foci with a pure plexiform pattern. C, Mild degree of scattered nuclear enlargement and hyperchromatism without apparent sarcomatous overgrowth should not be a source of concern in most cases. |
of diffuse NF is the recently reported neural ISF-like tumor because it expresses CD34 and S-100 protein and has a recurrent NTRK1 fusion.24
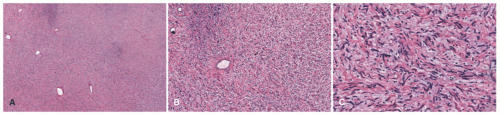 FIGURE 24-6. A-C, Neurofibroma, degenerative atypia. Such changes should not be viewed as an indication of malignancy. |
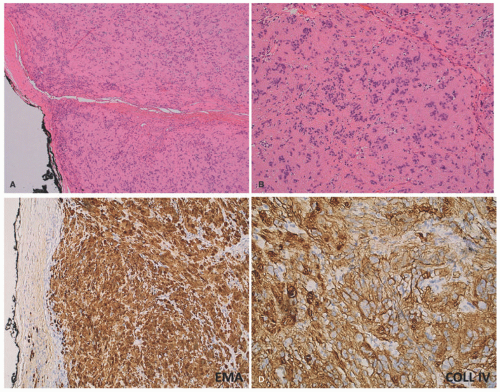 FIGURE 24-7. A-D, Schwannoma presenting on the upper eyelid of an 11-year-old male with NF2. A, The well-defined peripheral capsule is shown together with the inked margin. B, The fibrillary background is composed of cytoplasmic processes. The nuclei are arranged in small aggregates suggesting the formation of pseudorosettes. C, Unlike the neurofibroma, S100 protein is strongly and diffusely positive. D, Collagen type IV immunostain demonstrates the characteristic pattern of basement membrane positivity. |
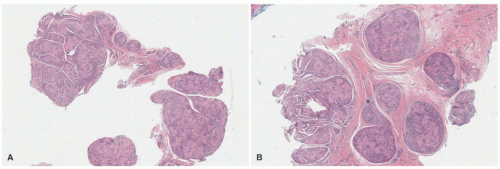 FIGURE 24-8. A-D, Plexiform schwannoma. Numerous plexiform profiles are composed of uniform spindle cells with focal Verocay body formation. Antoni A and Antoni B patterns are seen. Digital slides courtesy of Path Presenter.com. |
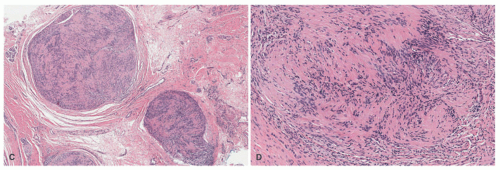 FIGURE 24-8. (continued) |
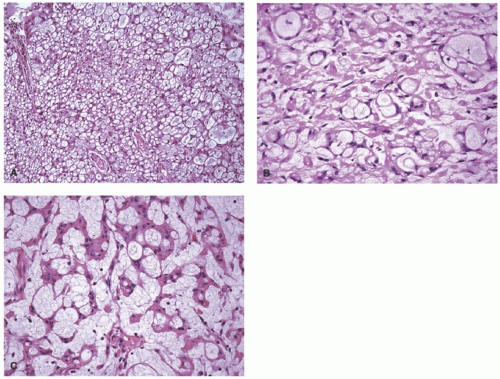 FIGURE 24-9. Myxoid-reticular pattern in schwannoma. Microcystic/reticular schwannoma. Views from three representative cases, highlighting the characteristically microcystic or reticular growth patterns. Obtained with permission Liegl B, Bennett MW, Fletcher CD. Microcystic/reticular schwannoma: a distinct variant with predilection for visceral locations. Am J Surg Pathol. 2008;32(7):1080-1087. |
unavoidable consideration, but in the context of a child, it is very unlikely. Schwannoma may have a predominant epithelioid cellular composition or may resemble a neuroblastoma.46,47
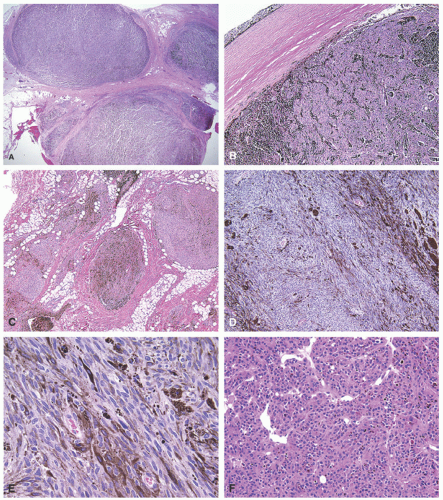 FIGURE 24-10. Psammomatous melanotic schwannoma. A, At low-power magnification, melanotic schwannoma typically grew as multinodular, generally circumscribed masses. A minority of cases showed at least partial encapsulation (B), but more often the tumors were unencapsulated and grew into surround fibroadipose tissue (C). D, At higher-power magnification, the tumors were most often composed of a fascicular proliferation of melanin-containing, moderately variable spindle cells, with an inconspicuous vasculature. E, Spindled melanotic schwannoma, showing elongated nuclei with small nucleoli. F, A minority of tumors were composed largely of epithelioid cells, resembling cutaneous melanoma. Obtained with permission Torres-Mora J, Dry S, Li X, Binder S, Amin M, Folpe AL. Malignant melanotic schwannian tumor: a clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am J Surg Pathol. 2014;38(1):94-105. |
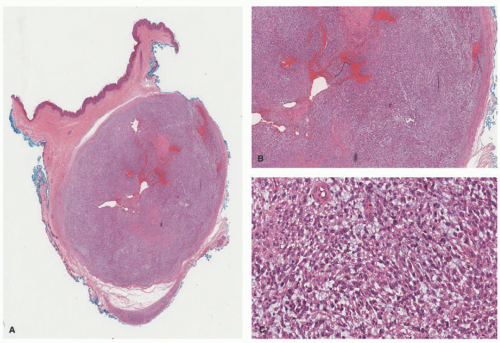 FIGURE 24-11. A-C, Epithelioid schwannoma. A single cell pattern with the features of a classic schwannoma focally in those tumors with trabecular to nodular profiles of epithelioid cells in a myxohyaline, myxoid, or dense collagenous stroma is the spectrum of microscopic features present in this tumor (A-C). Digital slides courtesy of Path Presenter.com. |
spindle cells. A myxoid background or plexiform growth may be prominent in some cases (Figure 24-12).2 Dense sclerosis is a feature in some cases. Epithelial membrane antigen reactivity is essential because the PN may also express CD34, smooth muscle actin, and S-100 protein (less than 10%).58
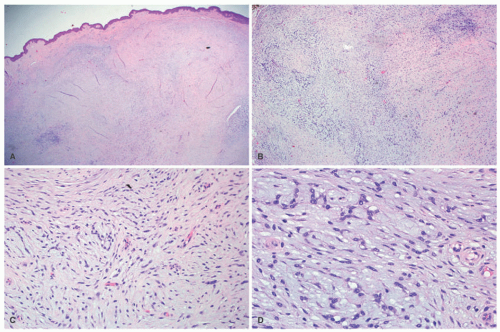 FIGURE 24-12. A-D, Perineurioma. Nodular and diffuse spindle cell tumor in the dermis that is well circumscribed on the edges (A and B). The lesion shows a storiform growth and myxoid background (C). Most of the cells show a cytoplasm with fibrillary features (D). Obtained with permission Dehner LP, Gru AA. Nonepithelial tumors and tumor-like lesions of the skin and subcutis in children. Pediatr Dev Pathol. 2018;21(2):150-207. |
the dermis, the small groups of granular cells blend into the background of dermal collagen (Figure 24-14). The nuclei of the tumor cells are rounded to ovoid with fine to coarse chromatin, but infrequently the nuclei may appear larger and hyperchromatic with an inconspicuous nucleolus. The granules are lysosomes that account for CD68 immunopositivity, in addition to characteristic S-100 protein reactivity (Figure 24-15). The congenital granular tumor or epulis has a densely cellular appearance as it forms a nodule or mass. With the rare exception, these tumors are nonreactive for S-100 protein.
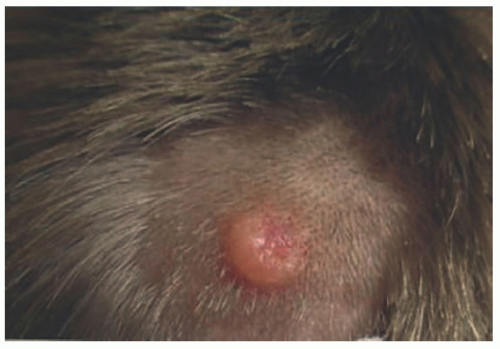 FIGURE 24-13. Granular cell tumor. Flesh-colored to slightly orange-hued soft nodule on the scalp of a young boy. Reprinted with permission from Olayiwola O, Hook K, Miller D, et al. Cutaneous granular cell tumors in children: case series and review of the literature. Pediatr Dermatol. 2017;34(4):e187-e190. |
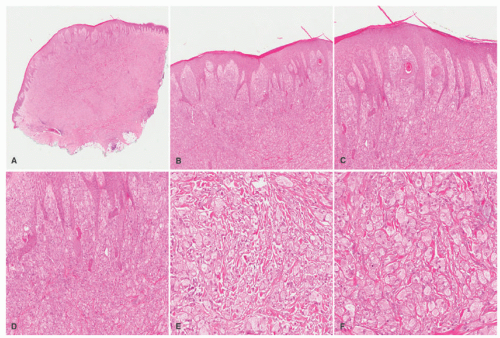 FIGURE 24-14. Granular cell tumor. Note the presence of pseudoepitheliomatous changes in the surface epidermis (A-C). Such changes can be confused with those of squamous cell carcinoma. Nested and diffuse sheets of uniform, pale rounded to ovoid to spindle shaped with abundant eosinophilic granular cytoplasm are the principal microscopic features of this tumor (D-F). Obtained with permission Dehner LP, Gru AA. Nonepithelial tumors and tumor-like lesions of the skin and subcutis in children. Pediatr Dev Pathol. 2018;21(2):150-207. |
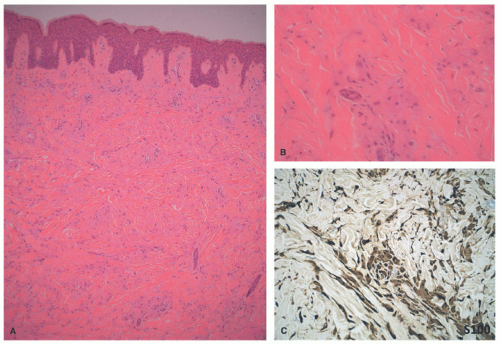 FIGURE 24-15. A-C, Granular cell tumor presenting in the skin on the shoulder of a 9-year-old female. A, The dermis at this magnification is seemingly unremarkable, which may be the initial impression in a granular cell tumor with a diffuse infiltrative pattern. B, Granular cells are infiltrating into superficial subcutaneous fat. C, S-100 protein immunostain demonstrates the striking number of granular cells in the dermis. |
schwannoma, the spindle cells are uniformly S-100 protein positive and a peripheral capsule, if present, is at least focally positive for epithelial membrane antigen.
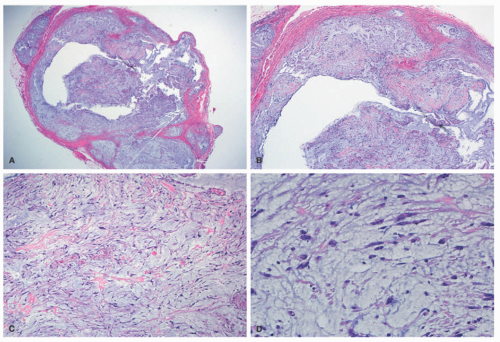 FIGURE 24-16. Dermal nerve sheath myxoma (myxoid neurothekeoma). A circumscribed, but nonencapsulated lesion is composed of multiple smaller nodules with a prominent myxoid or myomatous background containing dispersed spindle cells (A-D). Obtained with permission Dehner LP, Gru AA. Nonepithelial tumors and tumor-like lesions of the skin and subcutis in children. Pediatr Dev Pathol. 2018;21(2):150-207. |
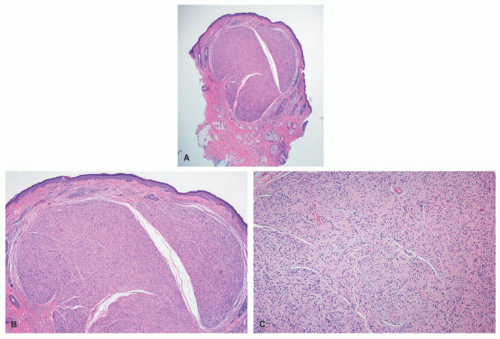 FIGURE 24-17. Palisaded encapsulated neuroma. Solitary well-circumscribed nodule with a schwannian appearance is present in the dermis (A-C). Obtained with permission Dehner LP, Gru AA. Nonepithelial tumors and tumor-like lesions of the skin and subcutis in children. Pediatr Dev Pathol. 2018;21(2):150-207. |
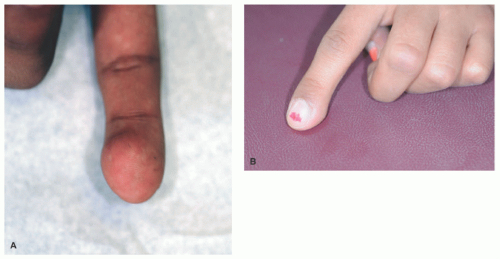 FIGURE 24-18. Traumatic neuroma. Nodule in the finger (A and B). |
of the traumatic neuroma occurs after ligature amputation of a rudimentary digit in polydactyly, whereas this complication is avoided with surgical excision.84,85
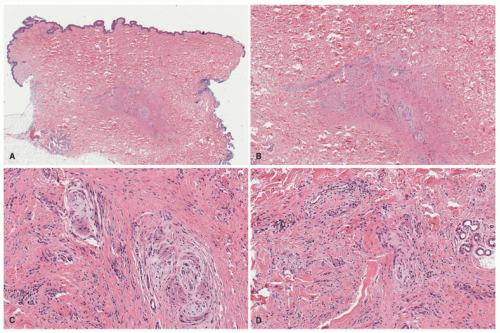 FIGURE 24-19. A-D, Traumatic neuroma. A background of fibrosis (scar) contains numerous small peripheral nerve twigs in a more or less circumscribed focus. A mixture of axons, Schwann cells, and perineurial cells is the composition of the neuroma. Digital slides courtesy of Path Presenter.com. |
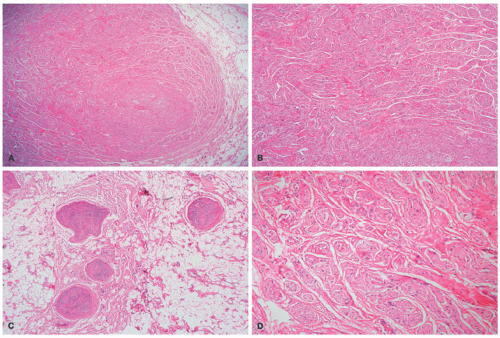 FIGURE 24-20. Neural fibrolipomatous hamartoma. Lobulated adipose tissue is evident and individual nerve bundles with perineural spindle cell proliferation composed of CD34 perineurial cells are encompassed by adipose tissue (A and B). Several contiguous nerve bundles may be surrounded by mature fat around the periphery of these nerves (C and D). Obtained with permission Dehner LP, Gru AA. Nonepithelial tumors and tumor-like lesions of the skin and subcutis in children. Pediatr Dev Pathol. 2018;21(2):150-207. |
masses have a predilection for the head and neck region, but not exclusively so.90 Those lesions presenting in the skin and subcutis often come to the attention of the pediatrician and dermatologist, whereas a nasal or oronasopharyngeal lesion is more likely seen by the pediatrician. All of these lesions have a common histogenesis of a presumed extracranial sequestration of neuroectodermal tissue.
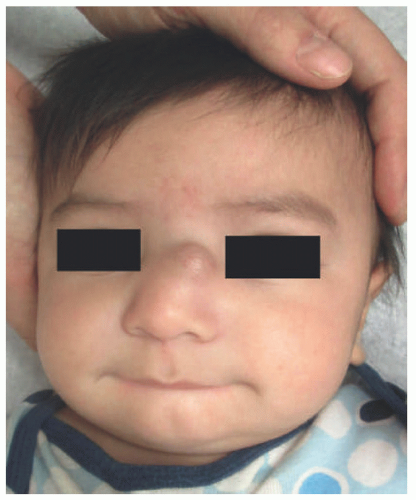 FIGURE 24-21. Nasal glioma. |
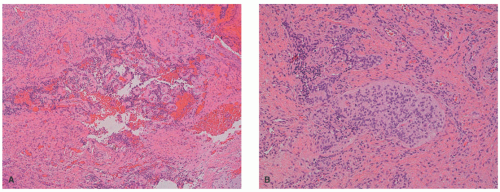 FIGURE 24-22. A, Myxopapillary ependymal rest presenting as an anal skin tag in a 2-day-old female. Note the myxohyaline vascular profiles surrounded in part by small primitive cells. A somewhat similar pseudovascular architecture is seen in the ectopic rudimentary meningocele. B, Another focus shows a circumscribed nodule of immature neuroglia. |
subcutis is the location of a circumscribed focus of collapsed, irregular spaces with an inconspicuous lining and/or a small nest(s) of compact, bland-appearing round cells, calcifications (psammomatous in some cases), and giant cells. A solid glial nodule may be present in some cases to provide a clue to neurogenic process of some kind. The giant cells and a pseudoinfiltrative pattern may suggest GCF or a vascular anomaly.112,113 The spaces are immunoreactive for vimentin and epithelial membrane antigen and nonreactive for CD31.
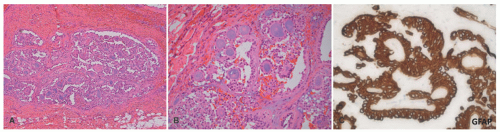 FIGURE 24-23. A-C, Myxopapillary ependymoma presenting as a “gluteal cyst” in a 5-year-old female. A, The tumor is composed of multiple circumscribed nodules of papillary profiles with central myxohyaline cores. B, The individual papillae are lined by low-grade cuboidal cells. C, Glial fibrillary acidic protein (GFAP) immunostaining shows strong diffuse positivity. |
One of the consistent histologic features if the biopsy includes the subcutis is the near total replacement of the dermis and infiltration of the subcutaneous fat (Figure 24-26). The latter finding is not specific to DFSP because it is also present in diffuse NF and some of the fibrous tumors in an infant or young child. The classic pattern of DFSP consists of uniform, compactly arranged, spindle cells in fascicles and/or storiform profile, which replace the dermis and extend in a confluent fashion into the subcutis (Figure 24-27A-C). Fusiform cells have uniform nuclei with minimal pleomorphism and sparing mitotic activity. In addition to this basic pattern, myxoid and/or collagen-rich foci may be seen as adjoining foci with the classic pattern or as the exclusive pattern in some cases. The presence of scattered multinucleated giant cells in clefted or pseudovascular spaces in a spindle cell background or loose fibrous stroma are the features of GCF (Figure 24-28A-C). The GCF pattern can be localized in a DFSP with classic or fibrous features, may represent the exclusive pattern, or is present as the initial histologic feature in a tumor that recurs with classic, myxoid, or fibrous features.113 Pigmentation of tumor cells is uncommonly present in DFSP in the so-called Bednar tumor. Immunohistochemistry is a useful adjunct to the diagnosis of DFSP as long as it is understood that other dermal and subcutaneous spindle cell tumors are CD34+.122 Typically, CD34 staining is uniformly and diffusely positive (Figures 24-29 and 24-30), but other markers have been reported to facilitate the diagnosis including CD99, D2-40, and apolipoprotein
D.121,123,124,125,126 Fluorescent in situ hybridization (FISH) is the most specific and sensitive method for the diagnosis.115
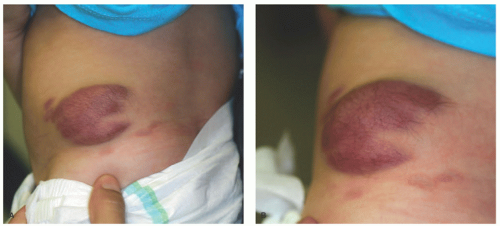 FIGURE 24-24. Dermatofibrosarcoma protuberans. Erythematous nodule on the trunk. |
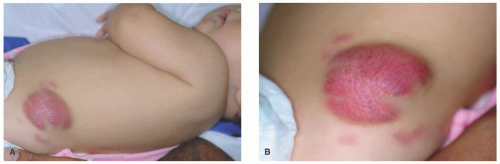 FIGURE 24-25. Dermatofibrosarcoma protuberans. Tumor on the flank. |
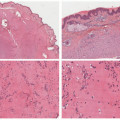
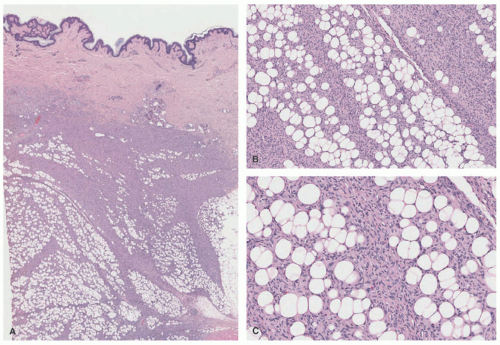 FIGURE 24-26. A-C, Dermatofibrosarcoma protuberans. Fibrohistiocytic proliferation with near total replacement of the dermis and infiltration of the subcutaneous fat (A). Uniform, compactly arranged, spindle cells in fascicles and/or storiform profiles that replace the dermis and extend in a confluent fashion into the subcutis (B and C). Digital slides courtesy of Path Presenter.com. |
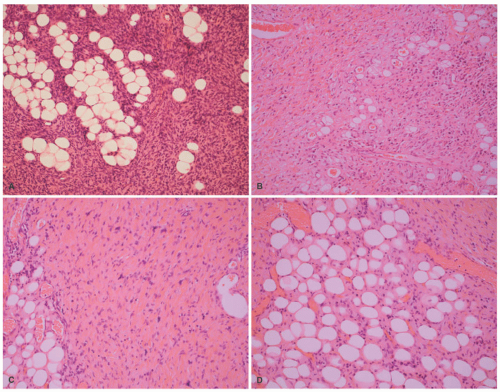 FIGURE 24-27. A-D, Dermatofibrosarcoma protuberans on the trunk of a 4-year-old male. A, Infiltration of tumor into the subcutis shows the overgrowth pattern. B, Another focus of this tumor shows a similar overgrown pattern, but note its more fibrous, hypocellular appearance. C, This focus shows scattered, enlarged tumor cells. D, The so-called honeycomb pattern of tumor growth is present in this focus. |
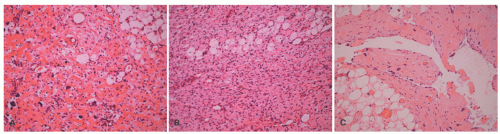 FIGURE 24-28. A-C, Giant cell fibroblastoma (GCF) presenting in the breast of a 7-year-old female. A, Numerous multinucleated giant cells with pericellular clear space are accompanied by a neoplastic fibrous stroma. B, Another area in the same neoplasm shows a diffuse fibrous pattern of this tumor. Most GCFs arise in a similar fibrous pattern. C, This GCF from the trunk of a 4-year-old male shows the exaggerated pseudoangiectoid pattern. |
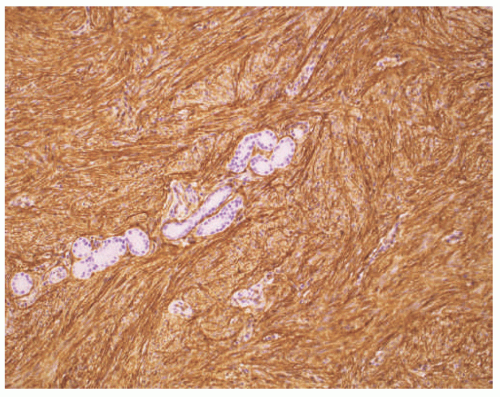 FIGURE 24-29. Dermatofibrosarcoma protuberans on the trunk of a 4-year-old male. Diffuse immunostaining for CD34 is characteristic of this neoplasm. |
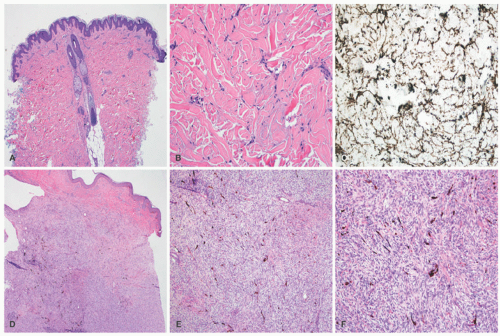 FIGURE 24-30. Dermatofibrosarcoma protuberans (DFSP) variants. The atrophic form of DFSP is particularly common in children and shows a very deceptive cytologic appearance (A and B). Diffuse CD34 staining (C) can be a clue to such diagnosis. The so-called medallion dermal dendrocyte hamartoma has identical histologic findings but lacks the PDGFB rearrangement seen in more than 90% of cases of DFSP. Pigmented DFSP (Bednar tumor) has the presence of deeply pigmented melanocytic cells admixed with the classic tumor cells of DFSP (D-F). Obtained with permission Dehner LP, Gru AA. Nonepithelial tumors and tumor-like lesions of the skin and subcutis in children. Pediatr Dev Pathol. 2018;21(2):150-207. |
was 13 years. Approximately 2% to 5% of DFs are diagnosed in the first two decades of life. A well-circumscribed, symmetric, round to ovoid, firm dermal nodule measures 2 cm or less in most cases.135 On occasion, the nodule may be accompanied by tenderness or pain.136 The overlying skin is often reddish-brown, shiny or scaly or darkly pigmented on the basis of so-called inductive changes in the epidermis.137,138 A central dimple on lateral compression, the “button hole sign,” is present in many cases. Multiple lesions are found in a small minority of cases where it was seen in 2% of children in our own series. Multiple DFs may present as congenital lesions, as eruptive nodules, or in children with trisomy 21.139,140
TABLE 24-3 Anatomic sites of dermatofibroma in 115 patients less than 20 years old | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
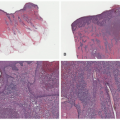
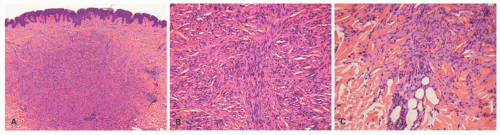 FIGURE 24-31. A-C, Dermatofibroma presenting on the breast of a 17-year-old female. A, The spindle cell tumor is centered in the dermis as a circumscribed nodule. Mild acanthosis noted in the overlying epidermis. B, This focus shows the uniform spindle cells arranged in a storiform profile. C, The periphery demonstrates the insinuation of spindle cells into adjacent dermis with collagen trapping. Note the limited extension. |
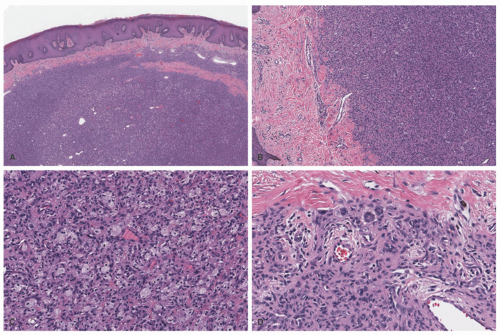 FIGURE 24-32. A-D, Dermatofibroma versus juvenile xanthogranuloma. Fibrohistiocytic lesion with epidermal induction changes (A). Collagen trapping at the periphery is seen (B). Xanthomatized cells (C) and Touton-type giant cells (D) are seen. Digital slides courtesy of Path Presenter.com. |
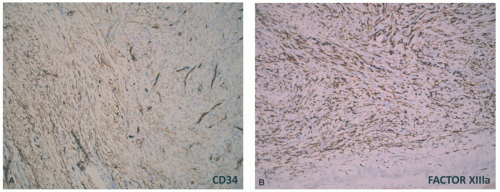 FIGURE 24-33. A and B, Dermatofibroma presenting on the back of a 10-year-old male. A, CD34 immunostaining demonstrates peripheral pattern of positivity, whereas only small blood vessels are highlighted in the body of the tumor. B, Factor XIIIa is diffusely positive within the tumor. |
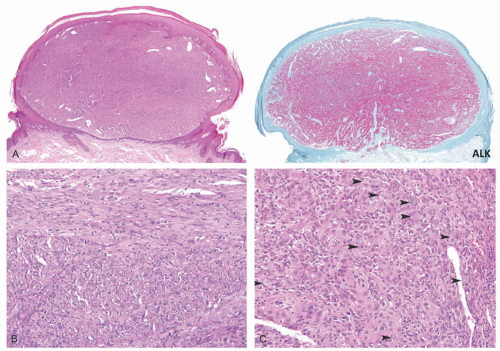 FIGURE 24-34. Epithelioid fibrous histiocytoma. Typical polypoid architecture (A) and composed of densely packed pale to clear spindle cells arranged in fascicles (B). Numerous mucinous cells are seen (C), arrow heads indicate the presence of mucinous cells. ALK is diffusely positive in the lesional cells. Adapted and obtained with permission from Kazakov DV, Kyrpychova L, Martinek P, et al. ALK Gene Fusions in Epithelioid Fibrous Histiocytoma: A Study of 14 Cases, With New Histopathological Findings. Am J Dermatopathol. 2018 Jan 11. |
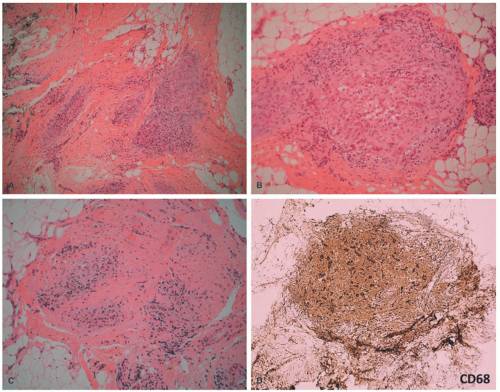 FIGURE 24-35. A-D, Plexiform fibrohistiocytic tumor presenting on the cheek of an 8-year-old male. A, Multiple nodules are present in a fibrous stromal background. B, The more cellular nodule has a granuloma-like appearance with a mixture of mononuclear and multinucleated cells. C, Here the nodules have a more fibrous appearance and may represent involution. D, CD68 immunostain shows intense positivity in the multinucleated cells. |
The radiating pattern of spindle cells in the subcutis has a resemblance to FHI, lipofibromatosis, and diffuse NF. The immature mesenchymal nodules of FHI and the spindle cells of lipofibromatosis and NF are CD34+ in contrast to the spindle cells of PFHT.22,23
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


