Abstract
This chapter focuses on key events and regulatory mechanisms that result in the morphogenesis, maintenance, and regeneration of the skin and its appendages. Understanding skin development provides essential insights into genetic skin diseases and facilitates the development of therapeutic approaches to acquired as well as inherited disorders. Maintenance and repair of the skin are dependent on skin stem cells with multiple independent progenitor pools that have diverse biological potential and regulatory mechanisms. Harnessing stem cells for therapy, either directly or by reprogramming them into pluripotent stem cells, holds great promise.
Keywords
skin development, skin differentiation, ectoderm, neural crest, mesoderm, hair development, skin maintenance, skin repair, epidermal stem cells, bulge stem cells
Introduction
Development of the human embryo is a complex process involving highly orchestrated cell movements, proliferation, death, and differentiation. This chapter focuses on key events and regulatory mechanisms that result in skin morphogenesis, maintenance, and regeneration. The spectrum of cutaneous abnormalities that can result from mutations in genes with critical roles in skin development is discussed. Ultimately, improved understanding of the pathways and signaling cascades that are disrupted in genetic skin disorders will aid in the development of therapeutic approaches for patients with acquired as well as inherited skin disorders.
Embryonic Origin of the Skin
The skin is composed of diverse cell types of both ectodermal (e.g. keratinocytes, melanocytes, Merkel cells, neurons) and mesodermal (e.g. fibroblasts, hematopoietic cells such as Langerhans cells, endothelial cells) lineages. To understand the origins of these cells, it is important to review the early stages of embryogenesis. Immediately after fertilization, cells divide rapidly, and by the end of the first week, the embryo begins to implant into the uterine wall. During the third week, the embryo undergoes gastrulation, a complex process resulting in the formation of the three embryonic germ layers: endoderm, mesoderm, and ectoderm.
During the next stage of embryogenesis, ectodermal cells commit to either a surface ectodermal or neuroectodermal fate. The surface ectodermal cells eventually differentiate into the keratinocytes of the embryonic epidermis, whereas the neuroectodermal cells invaginate to create the neural tube in a process called neurulation. As the neural tube forms, cells in its dorsal portion separate to form the neural crest. An important neural crest-derived cell in the skin is the melanocyte. Although Merkel cells were once believed to be neural crest derivatives, it has now been established that they descend from the epidermal lineage. The lineage of the dermis depends on the body site. The dermis (and other mesenchymal structures) of the face and frontal scalp are derived from the neural crest, whereas the dermis elsewhere is derived from the mesoderm. Knowing from which germ layer and lineage different cell types in the skin derive helps one to understand the pathophysiology of cutaneous disorders such as Waardenburg syndrome in which craniofacial dysmorphism and hearing impairment as well as pigmentary abnormalities reflect disrupted migration and survival of neural crest-derived cells.
An overview of key events in the development of skin and its specialized structures is shown in Fig. 2.1 .
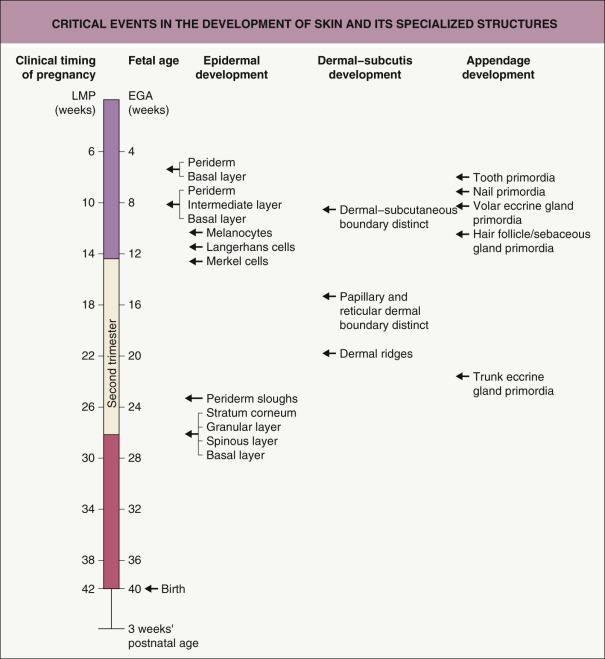
Epidermal Development
The ectoderm that covers the developing embryo after gastrulation is a single-layered epithelium ( Fig. 2.2A ). The first step in epidermal development occurs when cells of the surface ectoderm adopt an epidermal fate . Although this process does not result in major morphologic changes, it is marked by dramatic alterations in gene expression that result in the formation of the embryonic epidermis, which initially consists of a simple epithelium ( Fig. 2.2B ). Primitive keratinocytes subsequently generate cells of the periderm, a single cell layer that covers the developing epidermis until the cornified cell layer is formed ( Figs 2.2C–F & 2.3A,B ). The periderm is believed to exchange substances across fetal skin and to protect the developing epidermis from forming interepithelial adhesions.
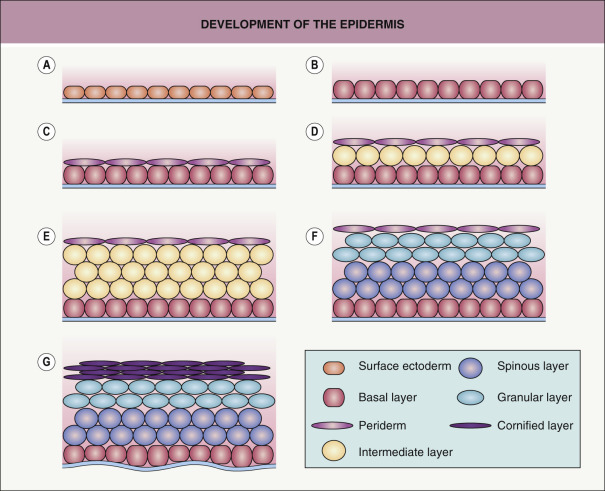
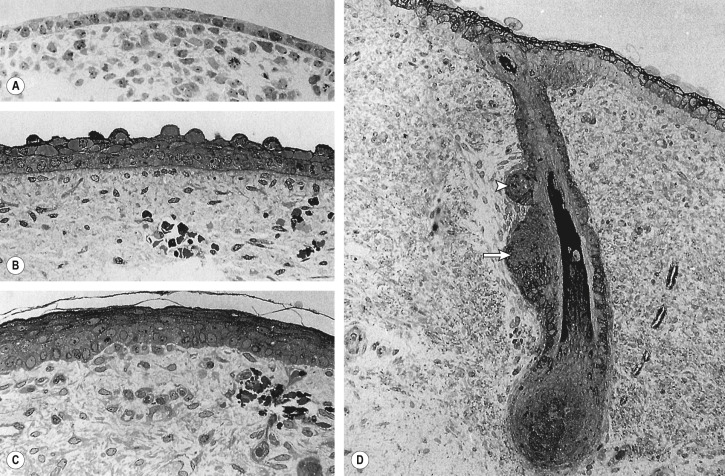
The embryonic epidermis begins to stratify at approximately 8 weeks’ estimated gestational age (EGA) . At this time, basic organogenesis is complete and bone marrow hematopoiesis commences, marking the transition from embryo to fetus. Of note, expression of TP63 is required for epidermal stratification. During the first stage of stratification, an intermediate cell layer is formed between the basal layer and periderm (see Figs 2.2D & 2.3B ). Unlike suprabasal keratinocytes in the postnatal epidermis, the intermediate layer consists of actively proliferating cells. As a consequence, it is able to expand to accommodate the rapid growth of the embryo as well as create additional layers of intermediate cells over the next several weeks (see Fig. 2.2E ). However, the intermediate cell layer is ultimately replaced by post-mitotic keratinocytes undergoing terminal differentiation.
Terminal differentiation, the process resulting in the formation of mature keratinizing epidermal cells, begins during the second trimester. Early cornification can be observed within the hair canal at approximately 15 weeks’ EGA , but it does not commence in the interfollicular epidermis until 22–24 weeks’ EGA, occurring first in the skin on the head, palms, and soles. The process begins when cells in the intermediate layer permanently withdraw from the cell cycle and differentiate into spinous and granular cells (see Fig. 2.2F ). The cornified cell layer, which is composed of “dead” keratinocytes (corneocytes) held together by a matrix of proteins and lipids (see Chs 56 & 124), subsequently starts to form and is several cells thick by 24–26 weeks’ EGA. The corneocytes are a reflection of the closely regulated process of terminal differentiation that is required for normal functioning of the skin. At the time of keratinization, the periderm detaches from the underlying epidermis and is sloughed off into the amniotic fluid, with remnants contributing to the vernix caseosa that coats newborns. During the third trimester, the number of keratohyalin and lamellar granules as well as stratum corneum layers increases. By the mid third trimester, the epidermis is morphologically similar to adult skin ( Figs 2.2G & 2.3C ), although it does not acquire full barrier function until a few weeks after birth.
Clinical Relevance
Genetic abnormalities affecting various stages of epidermal morphogenesis have been found to underlie inherited skin disorders in humans. However, generalized abnormalities in epidermal specification, the process through which the surface ectoderm adopts an epidermal fate, have not been identified. It is likely that generalized epidermal defects would be incompatible with survival past the first trimester. Mosaic skin conditions that result in abnormalities of the epidermis and/or its appendages often have a distribution pattern that follows the lines of Blaschko, which are thought to represent the migration pathways of epidermal cells during embryonic development. Classic examples include epidermal nevi (due to postzygotic mutations in genes encoding the fibroblast growth factor receptor 3, phosphoinositide-3-kinase alpha subunit, Ras family members, keratin 1, or keratin 10) and sebaceous nevi (due to dominant postzygotic mutations in HRAS > KRAS ) (see Ch. 62 ).
There are a number of genetic disorders that result in abnormal epidermal differentiation and barrier formation. One clinical presentation of such conditions is a “collodion baby” born encased in a taut, shiny, transparent membrane that is formed by aberrant stratum corneum. After shedding the membrane, most of these infants manifest with lamellar ichthyosis or congenital ichthyosiform erythroderma, two forms of autosomal recessive ichthyosis that exist on a spectrum . However, following a collodion membrane, some patients develop completely normal appearing skin. The so-called “self-healing” collodion baby is an example of a dynamic epidermal phenotype that depends on environmental conditions. All of these outcomes can result from mutations in the same set of genes that encode proteins essential for formation of the epidermal barrier, including transglutaminase-1 (an enzyme that cross-links lipids to the cornified cell envelope; TGM1 ), lipid processing enzymes ( ALOXE3 , ALOX12B ), and lipid transporters ( ABCA12 ) (see Ch. 57 ).
More deleterious mutations in ABCA12 cause harlequin ichthyosis (HI), an especially severe disorder of cornification characterized by aberrant epidermal maturation. Patients with HI are born with a tremendously thick, armor-like shell of hyperkeratosis, severe ectropion and eclabium, and underdevelopment of the nose and ears. The extreme phenotype of HI highlights the importance of lipid transport into lamellar bodies for epidermal formation and function.
Abnormalities in the stratum corneum are present not only in infants with ichthyosis, but also in premature infants, especially those born before 28 weeks’ EGA. The immaturity of the stratum corneum results in impaired barrier function, which leads to an increased risk of infection, dehydration, and excessive absorption of topical medications or chemicals . Even healthy full-term infants do not attain full skin barrier function until 3 weeks of age. The structural features of premature skin and adult skin are summarized in Table 2.1 .
| COMPARATIVE FEATURES OF PREMATURE, NEWBORN, AND ADULT SKIN | |||
|---|---|---|---|
| Premature | Newborn | Adult | |
| Skin thickness | 0.9 mm | 1.2 mm | 2.1 mm |
| Epidermal surface | Vernix (gelatinous) | Vernix | Dry |
| Epidermal thickness | ~20–25 microns | ~40–50 microns | ~50 microns |
| Stratum corneum thickness | 4–5 microns | 9–10 microns | 9–15 microns |
| 5–6 cell layers | >15 cell layers | >15 cell layers | |
| Spinous cell glycogen content | Abundant | Little or none | Little or none |
| Melanocytes | High number of cells; few mature melanosomes | Similar number of cells to young adult; low melanin production | Numbers decrease with age; melanin production dependent on skin type, body area |
| Dermal–epidermal junction | All known adult antigens expressed; fewer and smaller desmosomes | Structural features and antigens similar to those of the adult | Well-developed adhesive structures; large number of antigens expressed |
| Papillary dermis | |||
| Present but not marked | Present but not marked | Marked |
| Small | Small | Small |
| Abundant | Abundant | Moderately abundant |
| Reticular dermis | |||
| Marked | Marked | Marked |
| Small | Small to intermediate | Large |
| Abundant | Moderately abundant | Sparse |
| Elastic fibers | Sparse; tiny with immature structure | Small size and immature structure; distribution similar to adult | Large in reticular dermis, small and immature in papillary dermis; form network |
| Hypodermis | Well-developed fatty layer | Well-developed fatty layer | Well-developed fatty layer |
Development of Specialized Cells Within the Epidermis
Two populations of non-keratinocyte cells – melanocytes and Langerhans cells – migrate to the epidermis during early embryonic development. Melanocytes are derived from the neural crest that forms along the dorsal neural tube. Melanocyte precursors migrate away from the neural tube within the mesenchyme beneath the primitive epidermis. They follow a characteristic trajectory, moving dorsolaterally and then ventrally around the trunk to the ventral midline, anteriorly over the scalp and face, and distally along the extremities. Cutaneous melanocytes also arise from Schwann cell/melanocyte precursor cells that migrate along nerves to the skin via a distinct ventral pathway .
Melanocytes can be identified within the epidermis at approximately 50 days’ EGA based on HMB45 immunostaining and their dendritic morphology. Epidermal melanocyte density is high early in embryonic development (~1000 cells/mm 2 ), and it increases further (~3000 cells/mm 2 ) as the epidermis stratifies (80–90 days’ EGA) and the appendages begin to develop; later in gestation, the density decreases and becomes similar to that of young adults (800–1500 cells/mm 2 ). However, epidermal melanin production does not begin until 3–4 months’ EGA, and melanosome transfer to keratinocytes is not seen until 5 months’ EGA. Even though all melanocytes are functional and in place at birth, the pigmentation of the skin increases over the first few months of life; this process is most pronounced in infants with darker skin phototypes.
Active melanocytes are also present throughout the dermis during embryonic development. Eventually, most of these dermal melanocytes migrate to the epidermis or undergo apoptosis. By the time of birth, dermal melanocytes have generally disappeared, with the exception of certain anatomic sites (head and neck, dorsal aspects of the distal extremities, and sacrococcygeal area), which correspond to the most common locations for dermal melanocytoses and blue nevi (see Ch. 112 ).
Langerhans cell precursors appear within the epidermis during the first trimester and are detectable as early as 40 days’ EGA. These cells can be distinguished by their characteristic dendritic morphology; expression of CD45, HLA-DR, and CD1c; and high levels of ATPase activity. CD1a, Langerin, and Birbeck granules are expressed by 13 weeks’ EGA. The density of Langerhans cells in fetal skin remains low early in gestation and increases to typical adult levels during the third trimester.
Merkel cells , highly innervated neuroendocrine cells involved in mechanoreception, are initially identified within the epidermis during the first trimester. These cells are detected as early as 8–12 weeks’ EGA in palmoplantar epidermis, and slightly later in interfollicular skin. Merkel cells are identified by the presence of cytoplasmic dense core granules and the expression of cytokeratin 20 and neuropeptides. These cells are found in the basal layer of the epidermis, are often associated with appendages and nerve fibers, and are particularly dense on volar skin. The developmental origin of Merkel cells has been the subject of longstanding controversy, but genetic fate mapping studies have demonstrated that mammalian Merkel cells are derived from an epidermal rather than neural crest lineage .
Clinical Relevance
Several inherited pigmentary disorders result from genetic defects that lead to abnormal migration and proliferation of neural crest-derived melanocyte precursors (melanoblasts). Piebaldism and Waardenburg syndrome are characterized by achromic patches of skin on the central forehead, central abdomen, and extremities. This distribution pattern reflects the failure of melanocyte precursors to survive, proliferate, or travel to the distal points of their embryonic migration pathway. A number of different causative genes leading to this phenotype have been identified, including those encoding transcription factors (e.g. microphthalmia-associated transcription factor [ MITF ], PAX3 , SOX10 , SNAI2 ) as well as membrane receptors and their ligands (e.g. endothelin-3, endothelin B receptor, KIT receptor) (see Ch. 66 ). The endothelin B receptor is found on common melanoblast/ganglion cell precursors in the developing neural crest, explaining both the Hirschsprung disease and the pigmentary abnormalities that are associated with defects in this receptor or its ligand.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


