Sjögren’s Syndrome: Introduction
|
Epidemiology
Sjögren’s syndrome (SS) is one of the most common rheumatic autoimmune diseases. SS affects predominantly women with a female to male ratio of 9:1. Women are most commonly diagnosed in their fifth or sixth decade, but it can affect individuals of any age and sex.
SS has a worldwide distribution. In most studies done mainly in Caucasian populations the estimated prevalence rate in the adult general population is between 0.1% and 0.8%. However, one study in the United Kingdom showed a prevalence rate of 3.3%, whereas another study in Japan showed only a 0.02% prevalence rate. The estimated annual incidence rate was consistently approximately 0.005% in several studies.1
Pathogenesis
The pathogenesis of SS is still largely unknown. In a genetically predisposed individual, various environmental factors, such as viral infections, may lead to epithelial-cell activation and a protracted inflammatory response with features of autoimmunity. Autoreactive lymphocytes and autoantibodies are considered important in this process, although the pathogenic role of any particular autoantibody is still undefined. Abnormal apoptosis may be important in several ways. First, increased apoptosis of epithelial cells may lead to functional defects and provide autoantigens, whereas the prolonged survival of B and T cells through upregulation of antiapoptotic signals may be involved in sustaining a self-perpetuating autoimmune process. Decreased apoptosis of lymphocytes may also contribute to the increased frequency of lymphoma in SS patients.
The role of genetic factors in SS was recognized in family studies where first-degree relatives of patients had an increased prevalence of SS.2 Such family clustering was further observed among first-degree relatives of individuals with anti-Ro/SSA antibodies regardless of their clinical diagnoses (SS or systemic lupus erythematosus or even healthy controls).3
There is a well-established association between SS or anti-Ro/SSA and anti-La/SSB antibodies with HLA class II genes.4 Genes other than those of the HLA loci may also be associated with an increased risk of disease. Associations with a number of cytokine gene polymorphisms, such as interleukin (IL) 6, IL-10, tumor necrosis factor-α (TNF-α) and the IL-1 receptor antagonist, have been reported, but none of these have been confirmed to date.4 Several genetic polymorphisms previously linked to systemic lupus erythematosus and other autoimmune diseases are also associated with SS. From these, two transcription factors, (1) signal transducer and activator of transcription 4 (STAT4)5 and (2) interferon regulatory factor 5 (IRF 5),6 which were both independently associated with SS showed an additive effect in increasing the risk of Sjögren’s syndrome from around 1.6–1.9 for one risk allele to 6.7 when both risk alleles were present.7
The inciting event in the pathogenesis of SS is not known, and it may not be a single event. The strong predominance of females suggests gender-specific predisposing factors. Although sex hormones are obvious targets, there is no conclusive proof yet that the difference in the pathogenesis between males and females is due to sex hormones alone.8,9 Viral infections have also been proposed as inciting events. This theory is strongly supported by the fact that chronic inflammation of the salivary glands has been observed with chronic hepatitis C and human immunodeficiency virus infections, and such infections cause a disease with a clinical spectrum very similar to SS. The fact that some viruses, such as Epstein–Barr virus (EBV), are known to replicate in oropharyngeal and lachrymal glands led to the hypothesis that these viruses may be involved in Sjögren’s pathogenesis. In fact, genetic material from EBV was detected by DNA hybridization in SS salivary tissue, but it was also found in normal individuals. Further experiments have identified an unusual strain of EBV containing a deletion in the genome in salivary gland specimens from Chinese SS patients.10 Similarly, in a Japanese cohort, defective human T-lymphotropic virus-I genome was isolated from salivary gland tissue.11 Other viruses, such as coxsackievirus12 or endogenous retroviruses have also been proposed as causative agents. However, there is no proof, to date, that any of these viruses play a pathogenic role in SS.
Sections of salivary and lachrymal glands in SS are characterized by periductal mononuclear infiltrates. The majority of the infiltrating cells are CD4+ T lymphocytes, whereas CD8+ cytotoxic T cells are found in smaller numbers. Activated B lymphocytes are also present, including immunoglobulin-secreting cells.
Inflammation seems to target glandular epithelial cells, and recent research has demonstrated the central role of epithelial-cell activation in initiating the recruitment of the inflammatory infiltrate.13 In animal models, changes in epithelial cells occur before the appearance of lymphocytes in the salivary glands. Initial events include the expression of HLA class II molecules and various activation markers, such as the costimulatory molecules CD80 and CD86, on the surface of acinar and ductal14 salivary epithelial cells. These molecules are crucial in regulating the interaction between antigen-presenting cells and lymphocytes. Their expression in acinar and ductal15,16 epithelial cells suggests that epithelial cells in SS act as antigen-presenting cells and actively participate in lymphocyte activation. Another important step is the upregulation of adhesion molecules and chemokines, all of which contribute to the recruitment of inflammatory cells, such as T and B lymphocytes, macrophages, and dendritic cells. The activation of lymphocytes leads to abnormal cytokine and chemokine expression,17 perpetuating the chronic inflammatory process characterized by a complex interaction between activated epithelial cells, the innate and acquired immune system. Extraglandular manifestations occur as a result of similar lymphocytic infiltrations at other organs. This is described by some as autoimmune epithelitis to better reflect the systemic nature of the disease. Some epithelial cells express Fas and Fas ligand18 and, as a consequence, undergo apoptosis; others may be destroyed by perforin, granzymes, and other cytotoxins produced by lymphocytes. However, in most patients, only partial destruction of the glands is noted. Local production of cytokines, autoantibodies, metalloproteinases, and other inflammatory mediators may contribute to the dysfunction of the remaining epithelial cells by various mechanisms. For example, it has been shown that TNF interferes with the intracellular trafficking of aquaporin-5, one of the water channels required for saliva production. Enhanced activity of the type-1 interferon system has been linked to multiple autoimmune diseases, including Sjögren’s syndrome. Increased expression of interferon-regulated genes was described both in the salivary glands and peripheral blood.19–21 One of the cytokines upregulated by interferon-α is B-cell activating factor (BAFF), which promotes B-cell survival and has also been found at higher levels in Sjögren’s patients.22,23 Interestingly, both of these cytokines can be upregulated by viruses.24
Autoantibodies are the hallmarks of systemic autoimmune diseases, including SS. The best-defined autoantibodies in SS are the anti-Ro/SSA and anti-La/SSB antibodies.25 Both are targeted against ribonucleoprotein antigens. Anti-Ro/SSA recognizes two RNA-binding proteins (the 52-kDa or the 60-kDa protein), whereas anti-La/SSB antibodies recognize RNA polymerase III. Anti-Ro/SSA antibodies are found in over 70% of patients with SS, but are not specific for SS and are frequently found in SLE and other autoimmune diseases even when there are no symptoms or signs of oral or ocular dryness. Anti-La/SSB is more specific; it is present in 50% of patients with primary SS or SS/SLE but is rarely seen in other diseases. The pathogenic role of these antibodies is not yet defined, but, because Ro and La are expressed on the surface of apoptotic epithelial cells, it is possible that an immune response against these antigens contributes to inflammation in the gland. The most compelling in-vivo evidence for a pathogenic role of these autoantibodies comes from newborns with fetal heart block born to women with anti-Ro/SSA and/or anti-La/SSB antibodies. These antibodies can cross the placenta and bind to Ro and La antigens located on the cell surface of fetal myocardial tissue, leading to fetal heart block. Other autoantibodies, such as antinuclear antibodies and rheumatoid factor, are frequently present in patients with both primary and secondary SS. Although they lack specificity, they are markers of a systemic autoimmune response and thus can help distinguish SS from other causes of salivary or lachrymal gland dysfunction.
In recent years, research has focused on identifying antibodies more specific for SS, such as anti-α-fodrin and antimuscarinic acetylcholine receptor antibodies, but the results have been controversial. The major stimulus for saliva production is the binding of acetylcholine to muscarinic acetylcholine receptors. The hypothesis that oral and ocular dryness could result from antibodies antagonizing the muscarinic acetylcholine receptor-3 is intriguing. These antibodies have been demonstrated to play an essential role in eliciting glandular dysfunction in the nonobese diabetic (NOD)mouse model of SS, possibly through an inhibitory effect on the receptor.26 In humans, however, results are still contradictory as multiple attempts to detect these antibodies with conventional immunologic methods have been fruitless.27,28
Clinical Manifestations
The dominating feature of SS is exocrine gland dysfunction, leading to the classic sicca symptoms of xerostomia (oral dryness) and xerophthalmia or keratoconjunctivitis sicca (dry eyes).
Oral dryness is the principal symptom of SS, caused by decreased saliva secretion, which is persistent and continuous throughout the day and night and can significantly compromise quality of life. Reduced salivation causes difficulty in chewing and swallowing dry foods. Dryness of the tongue and oral mucosa leads to an altered sense of taste and, at times, produces a burning discomfort, especially when eating acidic or spicy foods. Physical examination may reveal a red and fissured tongue with a characteristic atrophy of the filiform papillae or angular cheilitis. Ulcerations can be found, particularly in SS patients with dentures, usually in proximity to the mucosal surface that makes contact with the denture.
Saliva has antimicrobial properties, so lack of saliva can predispose to infections. Oral thrush is common and can be manifested as pseudomembranous or erythematous mucosal lesions. Furthermore, patients with SS have an increased incidence of caries. A characteristic feature of caries in SS is its primary location at the cervical and incisal regions of the teeth.
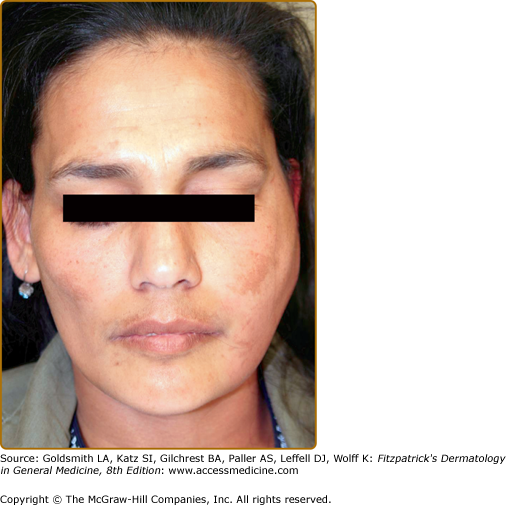
Bilateral salivary gland enlargement usually occurs in the parotid glands of SS patients. It is frequently nontender, and it can be recurrent or chronic (Box 161-1). Painful, unilateral parotid enlargement should raise the suspicion of an infection (Fig. 161-1) or a salivary gland stone. In cases of persistent unilateral parotid gland enlargement, the presence of lymphoma should be excluded (see Box 161-1).
Bilateral | Viral Infections | Mumps |
|---|---|---|
Epstein’s–Barr, cytomegalovirus, coxsackie | ||
Human immunodeficiency virus, human T-lymphotropic virus-I | ||
Hepatitis C | ||
Immune mediated | Sjögren’s syndrome | |
Sarcoidosis | ||
Amyloidosis | ||
Endocrine/metabolic | Diabetes mellitus | |
Hyperlipoproteinemia | ||
Chronic pancreatitis | ||
Acromegaly | ||
Other | Alcohol | |
Recurrent parotitis of the childhood | ||
Unilateral | Bacterial infections | |
Neoplasms | Mainly lymphomas | |
Sialolithiasis |
Medical causes of oral dryness, such as dehydration, diabetes, viral infections, or drug treatment, should be considered when evaluating a patient for Sjögren’s syndrome.
Ocular dryness is the other dominant feature of SS. A burning and itching sensation in the eyes, commonly exacerbated by smoke, is caused by lack of tear production. Patients frequently complain of intolerance to contact lenses. Paradoxically, the quantity of tears produced during crying may not be affected. Physical evaluation shows corneal injection and mucous discharge in the lower fornix. Enlarged lachrymal glands have been described in Sjögren’s patients, but occur less commonly than enlarged salivary glands. The constellation of symptoms and signs indicating dry eyes constitutes keratoconjunctivitis sicca.
Cutaneous xerosis, a term used to describe dryness of the skin, is very common in SS, with a frequency varying between 23% to 68%. The most common symptoms of xerosis are nonspecific pruritus, burning sensation and a pin-prick-like feeling. Physical examination reveals roughness, fine scaling, and loss of elasticity of the skin. The pathogenesis of xerosis is unknown. Impairment of the sweat glands is considered an important factor because decreased sweating has been reported in SS patients. A recent study has indicated that xerosis may be related to increased epidermal proliferation with disturbed epidermal differentiation.29
Dryness of the upper respiratory tract can cause epistaxis, hoarseness, and bronchial hyper-responsiveness. Another common complaint in women with SS is vaginal dryness, which may lead to an increased incidence of vaginal infections and dyspareunia.
Cutaneous manifestations are common extraglandular features of SS (Table 161-1).30 They are generally considered either vasculitic or nonvasculitic lesions.
Vascular |
|
Nonvascular |
|
Raynaud’s phenomenon is probably the most common abnormality. It can be seen in 15% to 35% of patients and can precede sicca symptoms by many years. Raynaud’s phenomenon in Sjögren’s syndrome is not accompanied by telangectasias as seen in systemic sclerosis. Calcifications have been described, but are uncommon. Although Raynaud’s is usually mild in SS, it is a marker of a subgroup with increased risk of extraglandular manifestations.31
Purpuric macules are very common in Sjögren’s. Flat, nonpalpable, blanching purpura has been associated with an entity called benign hyperglobulinemic purpura, characterized by polyclonal hypergammaglobulinemia and a positive rheumatoid factor. The skin biopsies reveal ruptured blood vessels with complement deposition.32
Cutaneous vasculitis (CV) can present as palpable purpura or urticarial vasculitis.
Palpable purpura, which does not blanch when pressure is applied to the skin is due to dermal vasculitis with extravasation of red blood cells, and typically involves the lower extremities and buttocks (see Chapter 164). It represents an important marker of more severe disease, and is associated with an increased risk of lymphoma development and mortality. Histopathologically, palpable purpurae can be divided into two groups. Neutrophilic inflammatory vascular disease is characterized by a predominantly neutrophilic infiltrate, fibrinoid necrosis, occlusion of the lumen, and extravasation of red blood cells, and is indistinguishable from the classical leukocytoclastic vasculitis (Fig. 161-2) (see Chapter 163). On the other hand, mononuclear inflammatory vascular disease is characterized by a mononuclear inflammatory infiltrate with invasion of the blood vessel walls. Fibrinoid necrosis is present but less prominent. The clinical presentation of these two forms are indistinguishable, but neutrophilic inflammatory vascular disease is associated more strongly with markers of systemic autoimmunity, such as antinuclear antibodies and anti-Ro/SSA and anti-La/SSB antibodies, hypergammaglobulinemia, rheumatoid factor, and hypocomplementemia. Cryoglobulinemic vasculitis can also be seen among Sjögren’s patients and has the same cutaneous manifestations (see Chapter 169).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


