Scleroderma: Introduction
|
Definition and Epidemiology
Systemic sclerosis (SSc) is a rare, multisystem disease, based on autoimmunological processes, vascular endothelial cell injury and an extensive activation of fibroblasts. It is characterized by a large individual variability in the extent of skin and organ involvement, as well as in disease progression and prognosis. The skin, esophagus, lung, heart and kidneys are the most frequently affected organs.
Women are more frequently affected by SSc, with a female-to-male ratio between 3:1 up to 14:1.1–4 The age of disease onset ranges between 30 and 50 years.4 However, male patients have earlier onset than female patients. Blacks with SSc are frequently younger than whites. Published data about incidence rates increased from 0.6 to 16 patients per million inhabitants, which is also true for the prevalence rates, which rose from 2 to 233 patients per million inhabitants per year,1–3,5 depending on methodological differences in case definition and ascertainment as well as the investigated time period.
SSc has the highest case-specific mortality of any of the autoimmune rheumatic diseases, but it varies individually depending on racial or ethnic differences, presence and severity of organ involvement, SSc subsets, age at diagnosis and gender differences. Although not curable, there have been substantial advances in treatment options for organ-based complications of SSc.
Etiology and Pathogenesis
The pathogenesis of this complex disease involves multiple cell types (endothelial cells, epithelial cells, fibroblasts, and lymphocytic cells) interacting through a variety of mechanisms that are dependent on their microenvironment and key mediators.
Major facets of the disease include inflammation, vasculature, and connective tissue-producing cells. The clinical heterogeneity of SSc makes it likely that distinct pathogenetic mechanisms predominate in particular patients or subsets of disease. Similarly, it is likely that the mechanisms are not the same at different stages of SSc. Although a genetic component to etiopathogenesis is likely and evidence supports genetic factors in severity and susceptibility, there is also strong evidence supporting environmental and chemical factors as triggers for the disease.
The best evidence for a genetic contribution to SSc comes from studies that report familial clustering and from the limited twin studies that have been undertaken. Although the absolute risk for familial occurring SSc is relative low, the relative risk for first-degree relatives is 13-fold higher compared to the normal population.6 Several studies suggest that a positive family history for SSc is the strongest risk factor, but ethnicity also contributes.6 Assassi et al suggest that members of SSc-affected families tend to show concordant scleroderma-specific autoantibodies.7 Studies of the whole-genome microarray expression studies of skin biopsies and circulating blood cells support the involvement of multiple and complex pathways in the development of SSc.6 Further support is provided from genetic association studies with candidate gene approaches. Most success has been observed in genetic analysis of individual components of the disease such as autoantibody profiles. These appear to have a strong genetic determinant, and this may underlie the apparent mutual exclusivity of the SSc hallmark reactivities. It has been demonstrated that the ability to mount an immune response to a particular SSc-associated antigen is restricted by major histocompatibility complex haplotype. Several studies suggest an association of HLA-DRB1*1302, DQB1*0604/0605 haplotypes with antifibrillarin positive patients,8 while HLA SRB1*0301 occurs in patients with anti-Pm-Scl antibodies.9 Observation from a large number of studies examining genetic markers has identified a number of candidate genes (AIF-1, CD19, CD22, CD86, CTLA-4, CCL-2, CCl-5, CXCL-8, CXCR-2, IL-1α, IL-1β, IL-2, IL-10, IL-13, MIF, PTPN22, TNF-α).6,10 However, as with other complex diseases, in very many instances, it has not always possible to replicate initially promising data. Studies of genetically homogeneous populations have been especially informative, including those of the Choctaw nation of Native Americans. However, it is of interest that some of the associations are very plausible in terms of molecular pathogenesis. It is likely that epistasis and the effect of multiple modifier genes confound simple genetic association studies in SSc, just as in other complex diseases.11
There is substantial evidence of inflammatory changes in the skin and lung of patients with SSc. One example is the presence of highly specific hallmark autoantibodies, which are summarized in Table 157-1. The first inflammatory infiltrates in lesional skin are predominantly cells of the monocyte lineage13 (T cells, macrophages, B cells, and mast cells). Later, T lymphocytes predominate and are detectable in both the circulation and affected organs. These T cells are predominantly CD4+, bear markers of activation, exhibit oligocloncal expansion, suggesting an antigen-driven proliferation, and show a predominant Th2-helper phenotype.14,15 Consequently, increased serum levels of Th2 cell-derived cytokines, i.e., IL-2, IL-4, IL-10, IL-13, and IL-17, have been observed in scleroderma patients.16,17 Besides T cells, B cells are also found in involved skin. Several studies suggest that B cells are able to induce extracellular matrix (ECM) production through secretion of IL-6 and transforming growth factor-β (TGF-β) and are involved in the production of autoantibodies.
Reactivity | Target Antigen | Frequency in SSc (%) | HLA Association | Clinical Association |
|---|---|---|---|---|
Centromere | CENP proteins speckled pattern | 20–30 | HLA-DRB1 HLA-DQB1 | Limited skin sclerosis, severe gut disease, isolated PAH, calcinosis |
Scl-70 | Topoisomerase-1 speckled pattern | 15–20 | HLA-DRB1 HLA-DQB1 HLA-DPB1 | Diffuse skin sclerosis, pulmonary fibrosis and secondary PAH, increased SSc-related mortality rate |
RNAP III | RNA polymerase III speckled pattern | 20 | HLA-DQB1 | Diffuse skin sclerosis, hypertensive renal crisis, correlated with a higher mortality rate |
nRNP | U1-RNP speckled pattern | 15 | HLA-DR2, -DR4 HLA-DQw5, -DQw8 | Overlap features of SLE, arthritis |
PM-Scl | Polymyositis/Scl nuclear staining pattern | 3 | HLA-DQA1 HLA-DRB1 | Limited skin sclerosis, myositis–sclerosis overlap, calcinosis |
Fibrillarin | U3-RNP nuclear staining pattern | 4 | HLA-DQB1 | Diffuse skin sclerosis, myositis, PAH, renal disease |
Th/To | 7–2RNP nuclear staining pattern | 2–5 | HLA-DRB1 | Limited skin sclerosis, pulmonary fibrosis |
Several of these autoantibodies are associated with defined subsets of the disease and are important diagnostic markers (see Table 157-1). The potential role of autoantibodies in pathogenesis is a fascinating and exciting area. The majority of SSc cases have circulating antibodies. These include a number of hallmark reactivities, as well as autoantibodies, that occur in other autoimmune rheumatic diseases (e.g., anticyclic citrullinated peptide, rheumatoid factor) but also antibodies that may have functional significance, since they are directed against cell surface antigens [antiendothelial cell antibodies (AECA), antifibrillin antibodies, anti-PDGF receptor antibodies, etc.]. However, functional impact of these antibodies remains an area of investigation. The best evidence of functional significance is for antiendothelial cell autoantibodies and for antifibroblast-reacting antibodies. Recent reports suggest the presence of antifibrillin autoantibodies and stimulatory autoantibodies reacting with platelet-derived growth factor (PDGF) receptors, although this observation requires further confirmation.
Microchimerism and graft-versus-host disease mechanisms have been suggested in some cases, although the relatively high frequency of microchimerism in healthy individuals or other disease states suggests that this may be contributory rather than causal if it has a role in SSc.
Vasculopathy in SSc is based on inappropriate vascular remodeling and repair processes. It involves the microcirculation and arterioles and is very likely a primary event in the pathogenetic processes of the disease and precedes fibrosis. Vascular abnormalities are characterized by vasoconstriction, adventitial and intimal proliferation, inflammation, and thrombosis.18 The earliest signs of vascular dysfunction are represented by enhanced vascular permeability with an imbalance between vasodilatory (NO, prostacyclin, calcitonin gene-related peptide) and vasoconstrictive mediators [endothelin 1 (ET 1), angiotensin II, α2-adrenoreceptors]. Consequently, the impaired blood-flow leads to tissue hypoxia, which induces strong expression of vascular endothelial growth factor (VEGF) and its receptors, associated with a defect of vasculogenesis. However, inflammatory cytokines like TNF-α may stimulate or inhibit angiogenesis depending on the duration of the stimulus.19
In addition to these functional abnormalities, intravascular and structural changes contribute to overt Raynaud phenomenon (RP), and in the course of time to progressive reduction of vessels and blood flow. These vasculopathies clinically manifest in all vessels of virtually all organs. Early lesions in the microcirculation because of structural damage are initially seen in the nail fold capillaries and as vasospastic responses in RP. Furthermore, vascular changes, i.e., overgrowth of the endothelium and deposition of scar tissue produce some of the major complications of SSc, including pulmonary arterial hypertension (PAH), scleroderma renal crisis (SRC), and digital vasculopathy.
SSc is a multisystem fibrotic disease. The initial inflammation and hypoxia in fibroblasts induces the production of several proteins that are involved in ECM remodeling as, for example, thrombospondin 1, fibronectin 1, lysylhydroxylase-2, TGF-β-induced proteins.20 Deposition of excessive ECM in specialized organs is responsible for much of the morbidity and mortality of the disease. Fibrous connective tissue is deposited by activated fibroblasts and myofibroblasts. From an early stage in the disease, an autonomous population of fibroblasts appears to be established that are responsible for the excessive production and accumulation of ECM. The initiators of this process include a number of key cytokines and growth factors that may represent logical therapeutic targets. The key factor seems to be a disturbed balance between synthetic and degradative mechanisms. Quiescent fibroblasts may be activated by TGF-β, connective tissue growth factor (CTGF), PDGF, or ET-1.21–24 There is increasing evidence that fibroblasts are induced to differentiate into myofibroblasts, which are characterized by a high contractility, ECM production, and cytokine release. Together with altered biophysical properties of the resulting connective tissue this leads to persistent activation of fibroblasts leading to the excessive deposition of ECM components.
A schematic summarizing pathogenetic mechanisms is shown in Fig. 157-1.
Figure 157-1
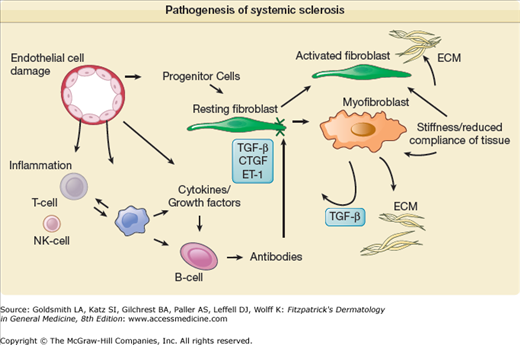
Pathogenesis of systemic sclerosis. The schematic shows how the development of systemic sclerosis results from a complex interplay between cells within the immune system, including adaptive and innate compartments, the vasculature, and the connective tissue. Cell–matrix interactions are important regulators of cellular functions. Early vascular events lead to later development of an autonomous population of activated fibroblasts and myofibroblasts that contract soft tissue and deposit excessive ECM. These cells may develop from resident connective tissue fibroblasts; transdifferentiation from other cell types, including activated microvascular pericytes; and recruitment of circulating progenitor cells (fibrocytes). The contribution of each lineage to the fibrotic lesion is still unclear. Many growth factors and cytokines are implicated as mediators of this process, and complex reciprocal networks may lead to a profibrotic microenvironment. Potential disease-modifying therapies could target individual mediators alone or in combination [e.g., tumor growth factor-β (TGF-β), endothelin (ET-1), connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF)] or modulate immune cells (e.g., cyclophosphamide) or the endothelial cell (e.g., prostacyclin analogues). The extracellular matrix is an important repository for mediators that are later released and play a key role in pathogenesis. CCL = CC chemokine ligand; Ig = immunoglobulin; IL = interleukin.
Scleroderma-like syndromes have been reported in association with numerous environmental toxins and drugs. These agents include solvents (vinyl chloride, benzene, toluene, epoxy resins), drugs (bleomycin, cardiopa, pentazocine, cocaine, docetaxel, metaphenylenediamine), and miscellaneous substances.25
SSc was reported to occur in underground coal and gold miners. In male patients with silicosis who were older than 40 years of age, the likelihood of developing SSc was approximately 190 times greater than in males not exposed to silica, and 50 times greater than in males without silicosis but exposed to silica dust.26 The role of silicone gel implants and other silicone products in the development of scleroderma has been questioned.27 However, most epidemiologic studies have failed to show a significant association. An unusual form of scleroderma characterized by RP, morphea-like skin changes, capillary abnormalities of the nail fold (similar to those in SSc), osteolysis of the distal phalanges, and hepatic and pulmonary fibrosis (PF) may occur in workers exposed to polyvinyl chloride. Bleomycin also produces PF, RP, and cutaneous changes indistinguishable from those of SSc.27 The development of these changes appears to be dose-dependent and is reversible on discontinuation of the drug. Collectively, chemical exposures account for a small fraction of scleroderma-like diseases. Large epidemiologic studies have not yet revealed a significant role for toxins and drugs in scleroderma.
Clinical Features of Systemic Sclerosis
The clinical manifestations of SSc depend to a large extent on the subset and stage of disease. The clinical features of established SSc are diverse, include severe fibrosis of the skin with all additional cutaneous manifestations and reflect the multiple patterns of internal organ involvement and the consequences of progression of the underlying pathologic processes of vasculopathy, inflammation, and fibrosis. Particular consideration must be given to the hallmark complications of hypertensive SRC, PAH, PF, and gastrointestinal (GI) dysmotility.
The heterogeneity of SSc arises from the range of disease manifestations that vary in extent and severity of organ involvement between patients. However, some clinical features that are almost always present are RP and skin sclerosis. The extent of skin sclerosis defines each major disease subset. Each subset has particular features, although there are common features to each.
The American College of Rheumatology (ACR) published in 1980 preliminary classification criteria for SSc to classify patients with established disease,28 showing 97% sensitivity and 98% specificity for SSc. The diagnosis is proven, if either one major criterion or at least two or more minor criteria are found. As major criterion are chosen scleroderma proximal to the metacarpophalangeal or metatarsophalangeal joints, and the minor criteria included sclerodactyly, digital ulcerations and/or pitting digital scars, and bibasilar PF.
Furthermore, in 1988 a descriptive subclassification of limited versus diffuse SSc by LeRoy,29 which was primarily associated with the extent of cutaneous involvement, has been widely accepted and used in clinical practice. In 2001, LeRoy and Medsger30 published amended criteria, with the additional presence of autoantibodies and nail fold capillaroscopic alterations. Furthermore, these criteria include a separate group of patients with early onset of SSc, with minimal skin thickening. It was mandatory that patients with early (limited) SSc have evidence of RP plus scleroderma-specific autoantibodies and/or nail fold capillaroscopic manifestations.31,32 However, there are several other classifications published, for example, by Nadashkevich O et al, Maricq and Valter, etc.32,33
Diffuse cutaneous SSc (dcSSc) is defined as a progressive form with an early onset of RP, usually within 1 year of onset of skin thickening. This subset is characterized by rapid skin involvement of trunk, face, upper arms, and thighs, showing very frequently, anti-Scl 70 (antitopoisomerase-I) or anti-RNAPIII antibodies.29 Furthermore, there is a higher propensity to develop PF, cardiac involvement, and SRC.
Limited cutaneous SSc is characterized by a long preexisting history of RP and skin changes of the extremities distal to the knee and elbow joints, including facial skin.29 This variant of SSc-subset often (50%–70%) presents with anticentromere-antibodies (ACA) and is frequently associated with isolated PAH. The traditional acronym CREST (calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasias) is nowadays obsolete and assigned to the limited form of SSc.
Patients with features of scleroderma together with those of another autoimmune rheumatic disease are combined; are designated overlap syndromes. This is defined as a disease occurring with clinical aspects of SSc (according to the ACR-criteria) or main symptoms of SSc simultaneously with those of other connective tissue diseases or other autoimmune diseases such as dermatomyositis, Sjögren’s syndrome, systemic lupus erythematosus, vasculitis, or polyarthritis. These patients present mostly high titers of anti-U1-RNP-, anti-nRNP-, antifibrillarin-, or anti-PmScl-antibodies.34
Patients suffering from early SSc, also known as undifferentiated SSc, are defined by positive RP and at least one further feature of SSc (positive nail fold capillary alterations, puffy fingers, pulmonary hypertension) and/or detectable scleroderma-associated autoantibodies without fulfilling the ACR-criteria.35
A very small proportion of cases (1.5%) develop vascular (RP and/or PAH), immunologic (most commonly anti-centromere antibodies), and organ-based fibrotic features of SSc but do not show skin sclerosis.36 Patients suffering from this subset are classified as SSc sine scleroderma.
In addition, the frequency and timing of different visceral manifestations of SSc differs between major subsets. However, there is some overlap between subsets in terms of organ-based disease and the extent and severity of skin sclerosis. In all patients, the extent and severity of skin sclerosis can be assessed by the modified Rodnan skin score (mRSS). Skin score at baseline correlates with disease severity and outcome in dcSSc. Thickening and fibrosis of the skin as one of the first recognized phenomenon in SSc still forms the basis of most classification criteria and proposed subsets of the disease.
Autoantibody-based classification of SSc has been proposed, and there is evidence from association studies that this may be clinically meaningful, as indicated in Table 157-1. Moreover, genetic association analysis using a candidate gene approach has demonstrated an association between serologically based subsets of SSc that are stronger than with SSc overall. The significance of this is uncertain, and it should be noted that a genetic basis for autoantibody reactivity has been well described, leading to the suggestion that the serologic subsets may be more genetically homogeneous than unselected SSc cases or clinically defined subsets of SSc.
Distinctive for this disease is the initial onset of RP, which appears in more than 90% of SSc-patients. It is defined by recurrent attacks of vasospasm of small digital arterioles/arteries at fingers and toes, usually caused by cold and/or other stimuli, for example, emotional stress. RP clinically appears suddenly and is clearly restricted and is accompanied by painful pallor/ischemia of single or several digits/toes, followed by reactive hyperemia after reheating at the end of a RP attack, in severe cases cyanosis (tricolore phenomenon) also ensues (see Fig. 157-2).
Figure 157-2
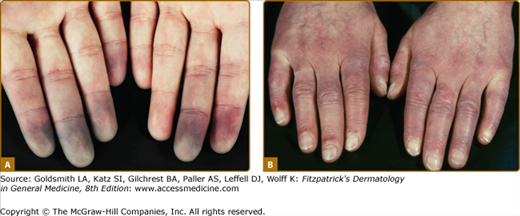
Clinical feature of patients with early disease. A. Raynaud phenomenon with typical discoloration (white–blue pallor), localized mostly at fingers and/or toes as the result of vasospasm. Coldness and emotional stress are the most frequently triggers for these attacks. B. Limited disease with puffy fingers.
Patients presenting only with RP should be studied for capillary alterations as well as autoantibody-status. To identify and visualize vascular-cutaneous alterations due to SSc, nail fold capillaroscopy is a noninvasive, simple, and one of the most useful diagnostic and prognostic methods (see Table 157-2). Furthermore, it is a useful tool to categorize capillary changes into early, active, and late patterns. Furthermore, laser Doppler perfusion imaging (LDPI) is also a noninvasive microvascular imaging technique able to provide maps of the cutaneous blood flow.37
Organ Involvement | Clinical Feature | Diagnostic Procedures |
|---|---|---|
Vascular system | Raynaud phenomenon |
|
Skin | Scleroderma Calcinosis cutis |
|
Musculoskeletal system | Arthralgia/Synovitis Muscle weakness |
|
Gastrointestinal tract | Reflux Dysphagia Diarrhoea, obstipation |
|
Respiratory system | Dyspnoea |
|
Cardiac system | Dyspnoea, arrhythmia |
|
Kidney | Renal function failure |
|
Skin involvement is a cardinal feature of SSc and usually appears first in the fingers and hands. Within time, patients develop nonpitting oedema of the fingers (puffy fingers) (see Fig. 157-2), hands, and extremities, whereupon an increasing induration and skin thickening appear (sclerodactyly). Depending on the localization of skin thickening, restricted mobility of joints (dermatogenous contractures), and/or restricted breath-excursion may be present. Typical facial features include telangiectasias, a beak-shaped nose as well as reduced mouth-aperture (microstomy). The typical physiognomy of FSSc patients is characterized by a radial furrowing around the mouth, no expression, a stiff and mask-like facial appearance, and sclerosis of the frenulum. Besides cosmetic/aesthetic problems, skin sclerosis causes considerable difficulties regarding eating and oral hygiene (summarized in Fig. 157-3).
Figure 157-3
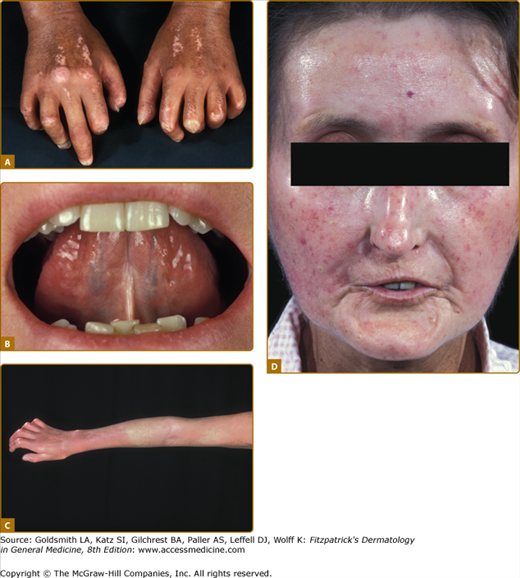
Extensive skin involvement in patients with dSSc. A. Sclerodactyly with dermatogenous contractures (restricted mobility of digital joints) as well as salt-and-pepper typical hyper- and hypopigmentations. B. Microstomia (radial furrowing around the mouth) with the typical frenulum sclerosis. C. Skin hardening/thickening proximal of the metacarpophalangeal-joints. D. Scleroderma-typical facial physiognomy with hypermimia, microstomia, telangiectasias and a beaked nose.
The abnormal deposition of cutaneous and/or subcutaneous calcium (calcinosis cutis), usually occurs over pressure points (acral, joints) (see Fig. 157-4). Calcinosis cutis next to joints is designated as Thieberge–Weissenbach syndrome. Further skin manifestations include hypo- and hyperpigmented skin (salt and pepper) (see Fig. 157-3), loss of hair follicles and sweat glands (hypo-/anhydrosis).
Figure 157-4
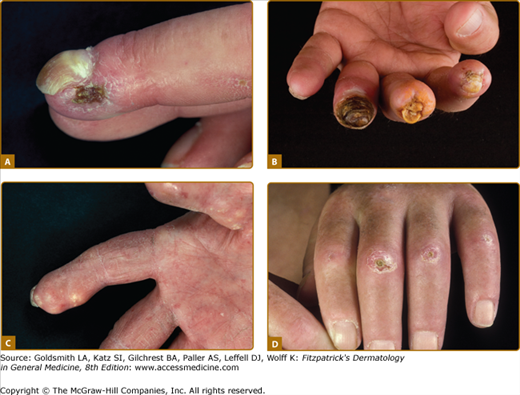
Digital alterations with complications. A. Digital ulcerations at the fingertips. B. Digital ulcerations and necrosis of the fingertips. C. Severe calcifications with deposition of subcutaneous masses. D. Multiple ulcerations at bone protuberantes with inflammation in the surrounding sclerotic skin.
Skin involvement/scleroderma should be evaluated using mRSS. Usually, 17 sites are assessed and skin thickness is categorized to grade 1, 2, or 3, corresponding to mild, medium, and severe, according to palpation of the skin by a trained examiner (see Fig. 157-5). Furthermore, new techniques for calculating skin thickening have been evaluated. Besides mRSS38 also 20-MHz ultrasound,39 MRI,40 and Plicometer41 are useful methods to assess skin thickening (recommended diagnostic procedures are listed in Table 157-2). Further physical procedures to monitor skin fibrosis are Durometer,42 Cutometer,43 and Elastometer.44 Besides all these noninvasive methods, a further appropriate but invasive method includes skin biopsy with histological evaluation of the dermal skin thickness. This also allows the characterization of the inflammatory infiltrates.
Figure 157-5
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


