Abstract
Purpura is a sign of several different processes including trauma, thrombocytopenia, anticoagulant use, cutaneous small vessel vasculitis, septic emboli and vascular coagulopathies, and is due to cutaneous hemorrhage. The number, distribution, and morphology of purpuric lesions are important factors in generating a probable and efficiently testable clinical hypothesis. Certain clinical features of the purpuric lesions, e.g. a retiform configuration, are critical in recognizing patients likely to have more serious systemic conditions.
Keywords
purpura, petechiae, ecchymoses, retiform purpura, pigmented purpura, Gardner–Diamond syndrome, capillaritis, palpable purpura, anticoagulants
Purpura
Introduction
The differential diagnosis of purpura is extensive, but the evaluation of a patient with purpura can be greatly facilitated by categorizing purpuric lesions into morphologic subsets . The number of lesions and their distribution pattern should also be assessed, including whether all the lesions are purpuric or just those located on the distal lower extremities.
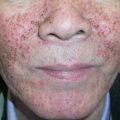
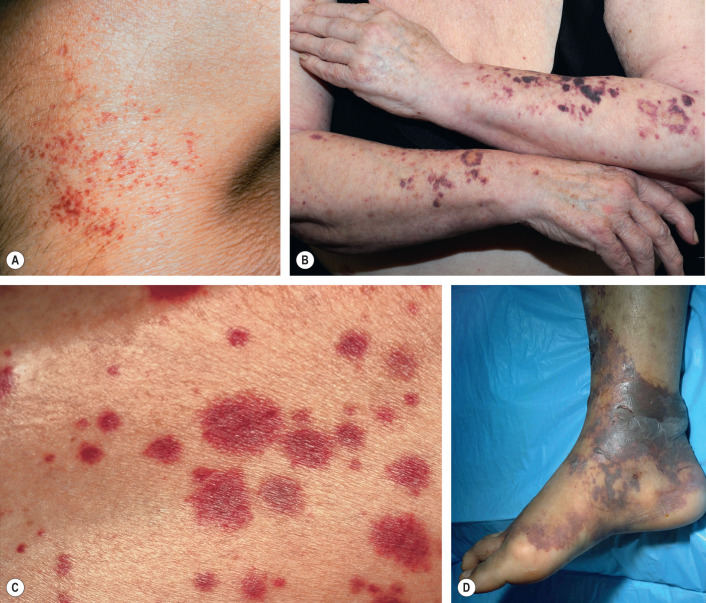
This chapter serves as an introduction to a method for evaluation and classification of patients presenting with purpura, defined as visible hemorrhage into the skin or mucous membranes. The differential diagnosis presented in this chapter is directed toward syndromes of primary purpura, where the hemorrhage is an integral part of lesion formation, rather than secondary hemorrhage into established lesions (e.g. stasis dermatitis, dependent cellulitis, dependent component of a morbilliform drug eruption). Purpuric lesions are divided into six subsets: (1) petechiae ( Fig. 22.1A , Table 22.1 ) ; (2) macular purpura ( Table 22.2 ); (3) macular ecchymoses ( Fig. 22.1B , Table 22.3 ); (4) palpable purpura ( Fig. 22.1C , Table 22.4 ); (5) non-inflammatory retiform purpura ( Fig. 22.1D ; Table 22.5 ); and (6) inflammatory retiform purpura ( Table 22.6 ). Lesions in the first three groups are categorized on the basis of size, while those in the last three groups can range in size from a few millimeters to several centimeters in diameter. The relevance of these morphologic subsets in sorting through the differential diagnosis is outlined in Fig. 22.2 .
| DIFFERENTIAL DIAGNOSIS OF MACULAR PETECHIAE (≤4 mm IN DIAMETER) |
| Hemostatically relevant thrombocytopenia (<10 000–20 000/mm 3 ) * |
| Major etiologies † |
|
| Abnormal platelet function |
| Major etiologies † |
|
| Non-platelet etiologies |
| Major etiologies † |
|
* Platelets provide factors necessary for hemostasis of endothelial cell junctions. When counts fall below 10 000–20 000/mm 3 , inadequate platelet support leads to degradation of junction function, increased permeability, and hemorrhage .
| DIFFERENTIAL DIAGNOSIS OF INTERMEDIATE MACULAR PURPURA (5–9 mm) |
| Major etiologies * |
|
| DIFFERENTIAL DIAGNOSIS OF MACULAR ECCHYMOSES (≥1 cm) |
| Procoagulant defect plus minor trauma |
| Major etiologies * |
|
| Poor dermal support of vessels plus minor trauma |
| Major etiologies * |
|
| Platelet disorders plus minor trauma |
| Major etiologies * |
|
| Miscellaneous |
|
| PALPABLE PURPURA: INFLAMMATORY PURPURA WITH PROMINENT EARLY ERYTHEMA |
| Leukocytoclastic vasculitis due to immune complex disease (see Fig. 24.1 ) |
| Small vessels only |
|
| Small- and medium-sized (macroscopic) vessels may be involved |
|
| Pauci-immune leukocytoclastic vasculitis (see Fig. 24.1 ) |
| ANCA-associated |
|
| Other |
|
| Not leukocytoclastic vasculitis |
| Small vessels only |
|
| Classic target lesion – usually erythema multiforme, but can be small vessel vasculitis, especially IgA-associated |
| DIFFERENTIAL DIAGNOSIS OF NON-INFLAMMATORY RETIFORM PURPURA |
| Occlusion primarily due to platelet-related thrombopathy |
| Major etiologies * |
|
| Cold-related precipitation or agglutination |
| Major etiologies * |
|
| Occlusion due to organisms growing in vessels |
| Major etiologies * |
|
| Systemic alteration in control of coagulation |
| Major etiologies * |
|
| Vascular coagulopathy |
|
| Embolization or crystal deposition |
| Major etiologies * |
|
| Reticulocyte, red blood cell occlusion |
|
| Miscellaneous |
|
** Also referred to as Coumadin ® necrosis.
^ Early lesions primarily thrombotic, but vasculitis can also occur.
| DIFFERENTIAL DIAGNOSIS OF INFLAMMATORY RETIFORM PURPURA |
| Vasculitis |
| Primarily dermal vessels |
|
| Dermal and subcutaneous vessels usually involved |
|
| Dermal vessel inflammation/occlusion/constriction |
|
Two major causes of purpura, microvascular occlusion syndromes and vasculitis, are discussed in Chapters 23 and 24 . The former are important to recognize because they may mimic vasculitis but require a very different approach to diagnosis and therapy. The most typical presentation of microvascular occlusion syndromes is non-inflammatory retiform purpura (see Table 22.5 ) . Early lesions seldom show much erythema, and in the uncommon instance in which early erythema is present, purpura or necrosis typically comprises at least two-thirds of the lesion.
In the skin, livedo reticularis is a reflection of the physiologic anatomy of slow flow states (see Ch. 106 ). It is the three-dimensional structure and flow regulation of the dermal and subcutaneous vasculature that gives rise to the net-like pattern of livedo reticularis. The diameter of the almost-circular individual units within the net-like grid varies from 2 cm or larger on the back to 5 mm or less on the palms or soles.
Retiform purpura morphology results from occlusion of the vessels that produce the livedo reticularis pattern, but the two entities can be distinguished based upon the presence or absence of purpura, respectively, hence the term “retiform purpura” . Given the size of dermal vessels, the clot within the vessel is often too small to be seen grossly. What is actually observed is hemorrhage around the vessel within the dermis, presumably due to ischemia with hemorrhage prior to complete occlusion of the vessel. The shape of such a hemorrhagic lesion is determined by the anatomy of the slow flow network, though a complete reticulate pattern is very rarely seen. Instead, the morphology of retiform purpura is composed of “puzzle pieces” of the livedo reticularis pattern. While some lesions have a branching appearance, very few are truly stellate, i.e. characterized by a central area of necrosis or hemorrhage with radiating extensions.
Retiform lesions should be carefully examined to confirm the presence of purpura. Inflammatory retiform purpura (see Table 22.6 ) may also have a reticulate or branching morphology, but it is accompanied by peripheral erythema. However, just as the wound healing response can eventually lead to erythema and leukocytoclasis in lesions initiated by non-inflammatory occlusion, vasculitis sometimes produces lesions that show minimal early inflammation ( Fig. 22.3 ). Among the inflammatory disorders that can result in retiform purpura, the ANCA-positive vasculitides (especially granulomatosis with polyangiitis and microscopic polyangiitis) are the ones most likely to present with lesions that are difficult to distinguish from retiform purpura due to occlusion. Despite this overlap of clinical presentations, careful examination of early lesions to determine whether there is a substantial inflammatory component can greatly aid in focusing the differential diagnosis and directing the evaluation.
This approach to the differential diagnosis of purpura represents a departure from traditional categorization by pathophysiology. Because the pathophysiology of purpuric syndromes is what the clinician is attempting to ascertain, sorting by pathophysiology is of limited clinical utility. A method based primarily on the morphology of purpuric lesions (in addition to number and distribution) is designed to streamline the process of generating clinical hypotheses and most likely diagnoses, thereby facilitating a rapid, efficient and accurate evaluation to prove or disprove the suspected diagnosis.
The Time Course of Purpura
The three subsets of macular (non-palpable) purpura (see Tables 22.1–22.3 ) typically develop via simple hemorrhage, with extravasated red blood cells and minimal inflammation observed in biopsy specimens. As such, these lesions have a very simple evolution, from initial hemorrhage to steady clearing of red blood cells and hemoglobin. Clinically, this correlates with fading of lesions and, in larger lesions, transition of color from red–blue or purple to green, yellow or brown before complete fading.
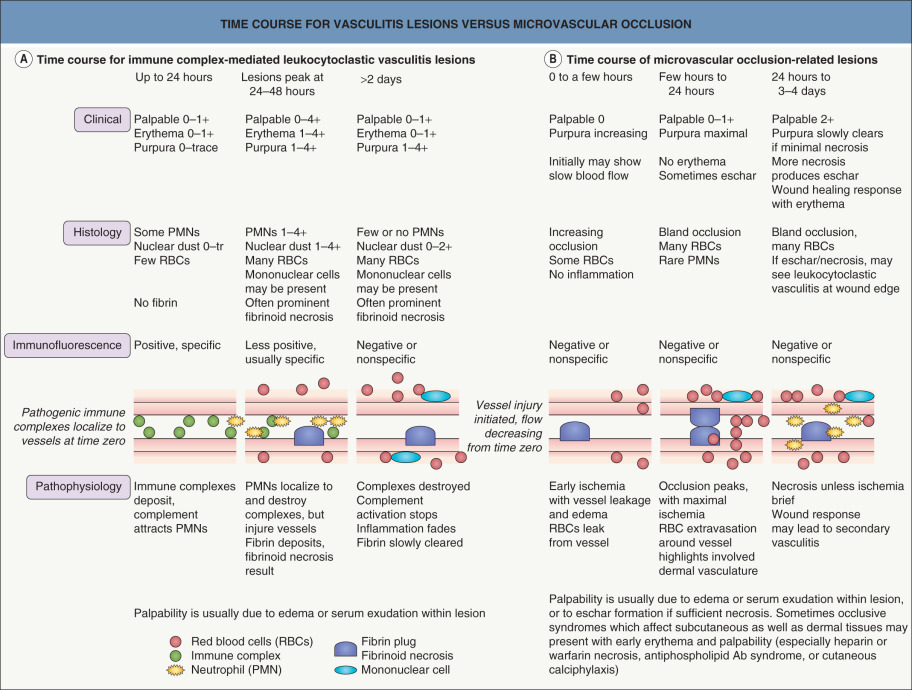
In syndromes of inflammatory hemorrhage, such as cutaneous small vessel vasculitis, as well as in microvascular occlusion, the evolution and clearing of lesions is more complicated. This is outlined in Fig. 22.3 . Because of the different evolution patterns of these two processes, it is possible for a biopsy specimen of a late lesion of vasculitis to resemble an early lesion of occlusion histologically. Conversely, a late lesion of occlusion with some resulting dermal necrosis may on histologic examination show features characteristic of leukocytoclastic vasculitis. This means that the biopsy specimen of a purpuric lesion must be interpreted within the context of the lesion’s age and clinical setting. The clinical and histologic assessments of purpuric lesions are equally important in properly evaluating and diagnosing a purpuric syndrome.
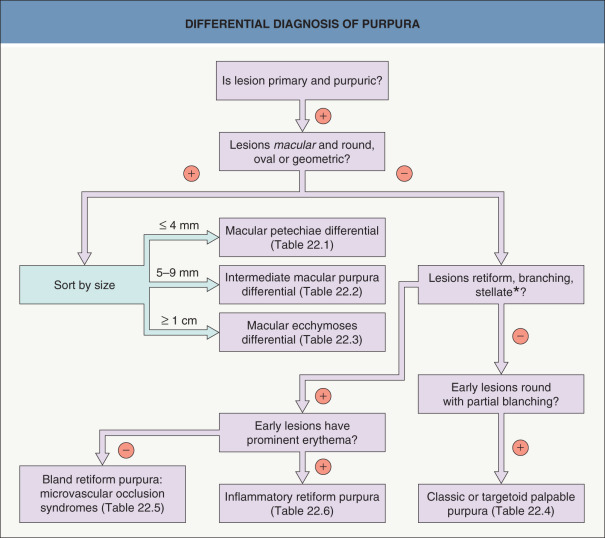
Coagulation
Primary hemostasis consists of the formation of a platelet plug, and this is sufficient for the many daily minor injuries to the microvascular system. When a platelet plug is inadequate due to the size of the vessel or the size of the injury, secondary hemostasis with clot formation is needed. The control of clot formation is critically important: too little clotting can lead to death by hemorrhage; inappropriate clotting produces thrombosis, embolus or necrosis; and uncontrolled clotting with fibrinolysis can produce both thrombosis and hemorrhage, as in disseminated intravascular coagulation. Therefore, a thrombus must form rapidly at sites of injury, but it should not extend beyond where it is needed, and clotting factors that escape the local injury site must be prevented from triggering clotting at distant sites. As might be expected for a system that requires exquisite regulation to perform properly, the control of coagulation is multifaceted and incompletely understood . Basal coagulation encompasses the constant low-level activation of some components of the procoagulant, natural anticoagulant, and fibrinolytic pathways, so that these systems are ready for a rapid response when needed. An overview of the controls and interconnections of these pathways is shown in Fig. 22.4 .