Porokeratosis: Introduction
|
Porokeratosis is a morphologically distinct disorder of keratinization, characterized clinically by hyperkeratotic papules or plaques surrounded by a thread-like elevated border that expands centrifugally. Histologically, a thin column of parakeratotic cells extends throughout the stratum corneum and is seen in all variants. This distinctive histopathologic feature, known as the cornoid lamella, corresponds to the raised hyperkeratotic border evident clinically.
At least six clinical variants of porokeratosis are recognized; however, the clinical distinction between these morphological variants may not be justified (Box 52-1). Reports of one type of porokeratosis coexisting with other forms and different types developing in multiple members of an affected family suggest more similarities than disparities, particularly in the disseminated forms.1–3
Porokeratosis of Mibelli | OMIM #175800 |
Disseminated superficial actinic porokeratosis (DSAP) | OMIM #175900 and #607728 |
Disseminated superficial porokeratosis (DSP) | |
Porokeratosis palmaris et plantaris disseminata (PPPD) | OMIM #175860 |
Punctate porokeratosis (PP) | |
Linear porokeratosis (LP) | |
Syndromic form: CAP (craniosynostosis, anal anomalies, and porokeratosis) syndrome | OMIM #603116 |
Etiology and Pathogenesis
Porokeratosis is a genetically heterogeneous disorder with multiple loci identified to date; however, the pathogenetic mechanisms remain elusive. Loci at chromosome bands 12q23.2–24.1 and 15q25 (DSAP1 and DSAP2) have been reported in familial disseminated superficial actinic porokeratoses; a further locus has been identified for disseminated superficial porokeratosis (DSP) at 18p11.3.4,5 The locus at DSAP1 corresponds to a candidate gene, SART3 (squamous cell antigen recognized by T cells 3); this encodes a tumor rejection antigen thought to be involved in the regulation of messenger RNA splicing. Fine mapping of the locus at DSAP1 has also revealed mutations in another potential candidate gene, SSH1 (slingshot 1), and a variation in the promoter region of ARPC3, which play a key role in actin dynamics.6–8 Missense mutations in SSH1 in this kindred have been shown to result in loss of heterozygosity in DSAP.9 However, microarray expression and real time quantative polymerase chain reaction (PCR) profiles of SART3, SSH1, and ARPC3 have failed to show differential expression patterns.10,11 Porokeratosis punctata palmaris et plantaris maps to a 6.9-centimorgan region at chromosome band 12q24.1–24.2, overlapping with the region identified for DSAP1, suggesting that the two forms may be allelic.12 The centrifugal expansion of lesions is postulated to reflect the migration of a mutant clone of keratinocytes.13 Supporting this mutant clone theory are findings of abnormal DNA ploidy and chromosomal abnormalities in lesional keratinocytes.14 The tumor suppressor proteins p53 and pRb are overexpressed in keratinocytes immediately beneath and adjacent to the cornoid lamella, although to date p53 mutations have not been identified, and there is no significant expression of p53 at an mRNA level.15–19,11 Cytogenetic abnormalities in fibroblasts, particularly on chromosome 3, have also been documented.20,21 Decreased mdm2, abnormal expression of psi-3, cytokeratins, filaggrin, and involucrin have also been reported.22,23 The increased prevalence of porokeratosis in immunosuppressed patients suggests that impaired immunity may be permissive in genetically predisposed individuals.24–27 Other reported triggering factors such as exposure to ultraviolet (UV) light, together with the increased potential for malignant transformation, highlight the dysplastic potential of affected keratinocytes. Malignant degeneration has been described in all variants of porokeratosis, with the exception of the punctate variety.28–30
Clinical Findings
Classic porokeratosis of Mibelli begins during infancy or childhood as asymptomatic small brown to skin-colored annular papules with a characteristic annular border (Fig. 52-1). The well-demarcated hyperkeratotic border is usually more than 1 mm in height, with a characteristic longitudinal furrow. The center of the lesion may be hyperpigmented, hypopigmented, depressed, atrophic, or anhidrotic. Lesions range in diameter from millimeters to several centimeters, but giant lesions measuring up to 20 cm may occur. Such giant porokeratoses are rare and occur predominantly on the lower leg and foot. Large lesions are associated with a higher malignant potential.30 Multiple lesions may arise; however, they are usually regionally localized and unilateral. The condition may be familial and inherited as an autosomal dominant trait. Lesions persist indefinitely.
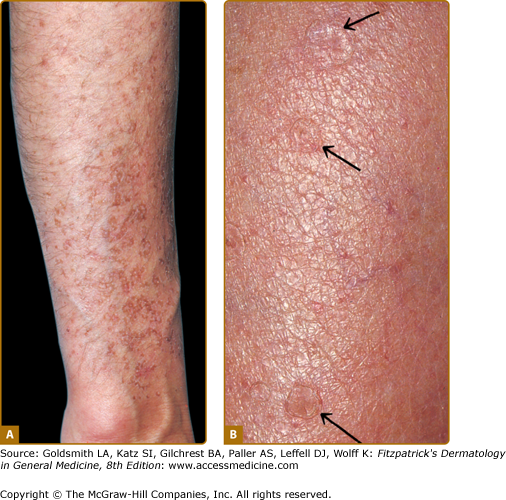
Disseminated superficial actinic porokeratosis (DSAP) is the most common of the porokeratoses. Lesions are characteristically uniformly small, annular, asymptomatic, or mildly pruritic papules ranging from 2 to 5 mm in diameter, distributed symmetrically on the extremities. Lesions are more generalized than other forms of porokeratosis, with typically in excess of 50 lesions located predominantly in sun-exposed sites (Fig. 52-2A and B). Although widespread, lesions typically spare palms, soles, and mucous membranes. Compared with porokeratosis of Mibelli, the hyperkeratotic border is characteristically more subtle. As the lesions progress, the older, central area becomes atrophic and anhidrotic. DSAP tends to be inherited as an autosomal dominant disorder, with the earliest reported age of onset at 7 years, and is usually fully penetrant by the third or fourth decade of life.5 Initial reports of induction of lesions by exposure to UV light and hypersensitivity of DSAP-derived fibroblasts to X-rays have not been consistently reproduced, and the pathogenesis of DSAP remains unknown.15,31–33
DSP also shows an autosomal dominant pattern of inheritance and has its onset in the third or fourth decade of life. Lesions primarily are morphologically identical to those of DSAP, occur on the extremities, and are typically distributed symmetrically, but do not spare sun-protected areas as in DSAP. As with DSAP, in excess of 100 lesions may be disseminated, with a predilection for the extensor surfaces of the extremities. Notably, involvement of the face is rare in both DSAP and DSP. In both disseminated forms, there is a reported female predominance, with a female–male ratio of 3:1.
Disseminated superficial porokeratosis in the context of immunosuppression is recognized following renal, hepatic, and cardiac transplantation, electron beam irradiation,34 immunosuppressive chemotherapy, and the use of systemic corticosteroids; with hematopoietic malignancies35; and in the setting of human immunodeficiency virus infection.36 Porokeratosis has also been reported after bone marrow transplantation in the absence of ongoing immunosuppressive therapy, which suggests a more complex association than immunosuppression alone.37 The distribution and morphology of DSP of immunosuppression are similar to those of DSAP, but a history of sun exposure is less evident.
Linear porokeratosis is an uncommon variant, traditionally categorized as a separate entity, but is increasingly recognized as mosaic manifestation of one of the other types of porokeratosis.38 Typically, it presents in early childhood, although congenital presentations have been reported.39 Two distinct clinical variants have been described. The more common presentation consists of a unilateral lesion confined to an extremity following Blaschko’s lines (Fig. 52-3). In the rare generalized form, multiple lesions affect several extremities and may involve the trunk. Linear variants have the highest potential for malignant degeneration of all the porokeratoses. It has been postulated that this association can be attributed to allelic loss due to a postzygotic mutation.40 The proband in Mibelli’s original publication most likely had coexistent linear porokeratosis and DSAP, with several subsequent reports confirming this phenomenon.4,38,41,42 These findings may be explained by the loss of heterozygosity for the DSAP allele and provides an example of the type 2 segmental manifestations of an autosomal dominant disorder.43,44
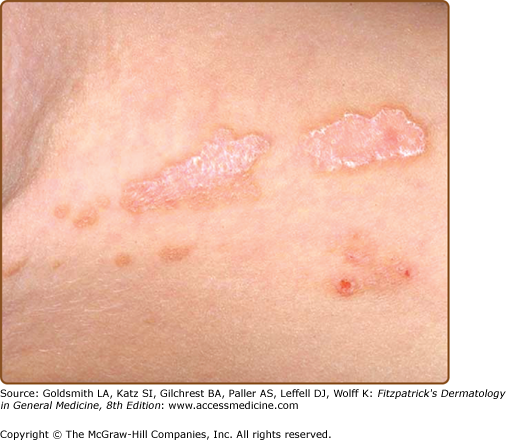
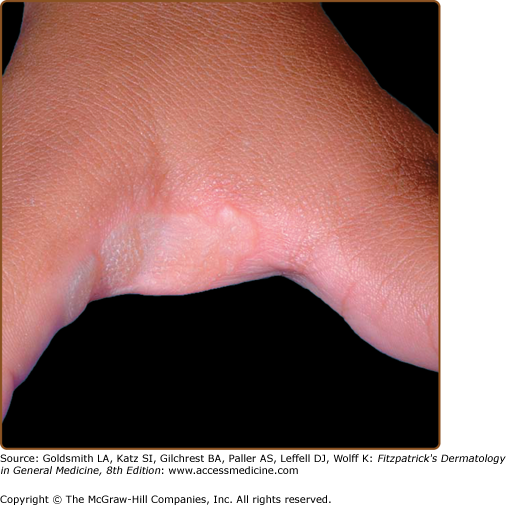
Porokeratosis palmaris et plantaris disseminata (porokeratosis punctata palmaris et plantaris) is a genodermatosis with an autosomal dominant inheritance pattern characterized by small, relatively uniform lesions (Fig. 52-4) that initially appear on the palms and soles. Subsequently, lesions spread to involve other parts of the body, including the mucous membranes and nonsun-exposed sites. The palmar and plantar lesions are generally more hyperkeratotic, and the characteristic longitudinal furrow along this ridge may be quite pronounced (Fig. 52-5). Typically, the lesions appear during adolescence or early adulthood and are bilateral and distributed symmetrically. Porokeratosis palmaris et plantaris disseminata affects males twice as often as females.
Figure 52-4