Chapter 8 Pathophysiology of burn shock and burn edema
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
Introduction and historical notes
Cutaneous thermal injury involving more than one-third of the total body surface area (TBSA) invariably results in the severe and unique derangements of cardiovascular function known as burn shock. Shock is an abnormal physiologic state in which tissue perfusion is insufficient to maintain adequate delivery of oxygen and nutrients and removal of cellular waste products. Before the nineteenth century, investigators demonstrated that after a burn, fluid is lost from the blood and blood becomes thicker; and in 1897, saline infusions for severe burns were first advocated.1,2 However, a more complete understanding of burn pathophysiology was not reached until the work of Frank Underhill.3 He demonstrated that unresuscitated burn shock was associated with increased hematocrit values in burned patients, which are secondary to fluid and electrolyte loss after burn injury. Increased hematocrit values occurring shortly after severe burn were interpreted as a plasma volume deficit. Cope and Moore4 showed that the hypovolemia of burn injury resulted from fluid and protein translocation into both burned and non-burned tissues.
Animal and clinical studies have established the importance of fluid resuscitation for burn shock. Investigations have focused on correcting the rapid and massive fluid sequestration in the burn wound and the resultant hypovolemia. The peer-reviewed literature contains a large experimental and clinical database on the circulatory and microcirculatory alterations associated with burn shock and edema generation in both the burn wound and non-burned tissues. During the last 50 years, research has focused on identifying and defining the mechanisms and effects of the many inflammatory mediators produced and released after burn injury.5
Burn shock is a complex process of circulatory and microcirculatory dysfunction that is not easily or fully repaired by fluid resuscitation. Severe burn injury results in significant hypovolemic shock and substantial tissue trauma, both of which cause the formation and release of many local and systemic mediators.6–8 Burn shock results from the interplay of direct tissue injury, hypovolemia, and the release of multiple mediators of inflammation, with effects on both the microcirculation and the function of large vessels, heart and lungs. Subsequently, burn shock continues as a significant pathophysiologic state, even if hypovolemia is corrected. Increases in pulmonary and systemic vascular resistance (SVR) and myocardial depression occur despite adequate preload and volume support.8–12 Such cardiovascular dysfunctions can further exacerbate the whole body inflammatory response into a vicious cycle of accelerating organ dysfunction.7,8,13 Hemorrhagic hypovolemia with severe mechanical trauma can provoke a similar form of shock.
Hypovolemia and rapid edema formation
Burn injury causes extravasation of plasma into the burn wound. Extensive burn injuries are hypovolemic in nature and characterized by hemodynamic changes similar to those that occur after hemorrhage, including decreased plasma volume, cardiac output, and urine output; and an increased systemic vascular resistance with resultant reduced peripheral blood flow.6,8,14–16 However, as opposed to a fall in hematocrit with hemorrhagic hypovolemia due to transcapillary refill, an increase in hematocrit and hemoglobin concentration will often appear despite fluid resuscitation. As in the treatment of other forms of hypovolemic shock, the primary initial therapeutic goal is to promptly restore vascular volume and to preserve tissue perfusion in order to minimize tissue ischemia. However, burn resuscitation is complicated not only by severe burn wound edema, but also by extravasated and sequestered fluid and protein in non-burned soft tissue. Large volumes of resuscitation solutions are required to maintain vascular volume during the first several hours after an extensive burn. Data suggest that despite fluid resuscitation normal blood volume is not restored until 24–36 hours after large burns.17
Edema develops when the rate by which fluid is filtered out of the microvessels exceeds the flow in the lymph vessels draining the same tissue mass. Edema formation often follows a biphasic pattern. An immediate and rapid increase in the water content of burn tissue is seen in the first hour after burn injury.15,18 A second and more gradual increase in fluid flux of both the burned skin and non-burned soft tissue occurs during the first 12–24 hours after burn injury.7,18 The amount of edema formation in burned skin depends on the type and extent of injury15,19 and on whether fluid resuscitation is provided, as well as the type and volume of fluid administered.20 Fluid resuscitation elevates blood flow and capillary pressure, thereby contributing to further fluid extravasation. Without sustained IV replacement of vascular fluid losses edema formation is somewhat self-limited, as tissue blood flow and capillary pressure decrease. Edema formation in thermally injured skin is characterized by an extremely rapid onset. Tissue water content can double within the first hour after burn.15,21 Leape found a 70–80% increase in water content in a full-thickness burn wound 30 minutes after burn injury, with 90% of this change occurring in the first 5 minutes.16,22,23 There was only a modest increase in burn wound water content after the first hour in non-resuscitated animals. In resuscitated animals or animals with small wounds, adequate tissue perfusion continues to ‘feed’ the edema for several hours. Demling et al.18 used dichromatic absorptiometry to measure edema development during the first week after an experimental partial-thickness burn injury on one hindlimb in sheep. Although edema formation was rapid, with over 50% occurring in the first hour, maximum water content was not present until 12–24 hours after burn injury.
Normal microcirculatory fluid exchange
An understanding of the physiologic mechanisms of the rapid formation of burn edema requires an understanding of the mechanisms of microvascular fluid balance. Under physiologic steady-state conditions blood pressure in capillaries causes a filtration of fluid into the interstitial space. Filtered fluid may be partially reabsorbed into the circulation at the venous end of capillaries and venules, but the bulk of the filtrate is removed from the interstitial space by lymphatic drainage.24–26 Fluid transport across the microcirculatory wall in normal and pathological states is quantitatively described by the Landis–Starling equation:
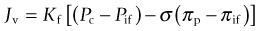
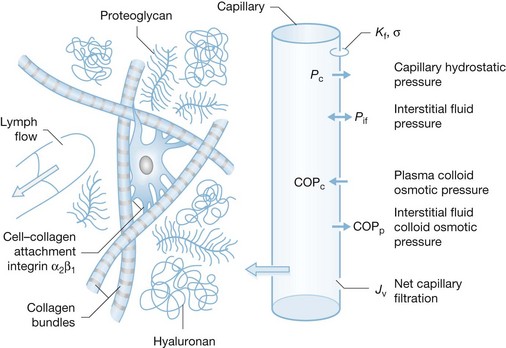
This describes the interaction of physical forces that govern fluid transfer between vascular and extravascular compartments. Jv is the volume of fluid that crosses the microvasculature barrier. Kf is the capillary filtration coefficient, which is the product of the surface area and hydraulic conductivity of the capillary wall; Pc is the capillary hydrostatic pressure; Pif is the interstitial fluid hydrostatic pressure; πp is the colloid osmotic pressure of plasma; πif is the colloid osmotic pressure of interstitial fluid; σ is the osmotic reflection coefficient. Edema occurs when the lymphatic drainage (JL) does not keep pace with the increased Jv (Fig. 8.1).
Mechanisms of burn edema
Capillary filtration coefficient (Kf)
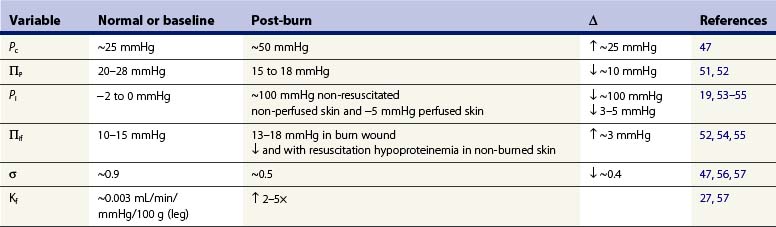
Burn injury causes direct and indirect mediator-modulated changes in the permeability of the blood–tissue barrier of the capillaries and venules. Arturson and Mellander27 showed that, in the scalded hindlimb of dogs, Kf immediately increased two to three times, suggesting that the hydraulic conductivity (water permeability) of the capillary wall increased. Kf is a function of both hydraulic conductivity and the capillary surface area. Thus, local vasodilation and microvascular recruitment contribute to the increased Kf in addition to increased hydraulic conductivity. Measuring Kf and the rate of edema formation (Jv) allowed Arturson and Mellander to determine the changes in transcapillary forces necessary to account for the increased capillary filtration. Their calculations indicated that a transcapillary pressure gradient of 100–250 mmHg was required to explain the extremely rapid edema formation that occurred in the first 10 minutes after a scald injury. They concluded that only a small fraction of the early formation of burn edema could be attributed to the changes in Kf and permeability. They further suggested that osmotically active molecules generating sufficiently large osmotic reabsorption pressures are released from burn-damaged cells. This hypothesis was never confirmed, and subsequent studies described below show that such large increases in filtration force could be attributed to increased Pc, and particularly to a large decrease in Pif (Table 8.1).
Capillary pressure (Pc)
In most forms of shock capillary pressure decreases as venous pressure decreases and less arterial pressure is transferred to the capillary due to arteriolar vasoconstriction. However, in studies using the vascular occlusion technique in the scalded hindlimb of dogs, Pc doubled from ~25 mmHg to ~50 mmHg during the first 30 minutes after burn injury, and slowly returned to baseline over 3 hours.47
Interstitial hydrostatic pressure (Pif)
An initially surprising, but now well-verified, finding is that Pif in dermis becomes extremely negative after thermal injury. Using micropipettes and a tissue oncometer, Lund19 reported that dermal Pif was rapidly reduced from its normal value of −1 mmHg to less than −100 mmHg in isolated non-perfused samples of skin. This large negative interstitial hydrostatic pressure constitutes a powerful ‘suction force’ or imbibition pressure, adding to the elevated capillary pressure in promoting microvascular fluid filtration. In vivo measurements show a temporary reduction of −20 to −30 mmHg; the less negative Pif in vivo is due to the continued tissue perfusion and fluid extravasation that relieves the imbibition pressure. After resuscitation, Pif was reported increased to a positive value of 1–2 mmHg in one study.48,49 On the other hand, Kinsky50 reported a continued negative pressure providing a partial explanation for the sustained edema for the first four hours post injury. During the first several days post burn injury tissue volume and hydration are greatly elevated, with a slight decrease or no change in Pif. By definition this implies an elevated interstitial compliance, which is likely to be the main mechanism that sustains burn edema.
The size of the decrease in Pif establishes it as the factor predominantly responsible for both the initial rapid development of edema and the sustained edema. The mechanism for the large decrease in Pif is due, at least in part, to the release of cellular tension exerted on the collagen and microfibril networks in the connective tissue via the collagen-binding β1-integrins. The integrins are transmembrane adhesion receptors that mediate cell–cell and cell–matrix adhesion, thereby allowing the glycosaminoglycan ground substance, which is normally underhydrated, to expand and take up fluid.58 The magnitude of the reduction in Pif is also observed in several non-burn inflammatory reactions (48/80 and PAF). However, these mediator-induced changes are milder, with a lowering of Pif to −5 to −10 mmHg. The much greater decrease in burn injury versus mediator-induced inflammation suggests additional mechanisms in burn edema. These mechanisms remain to be identified, but may result from the direct physical destruction of the connective tissues by heat.
Osmotic reflection coefficient (σ)
The osmotic reflection coefficient is an index of the proportion of the full osmotic pressure generated by the concentration gradient of plasma proteins across the capillary wall. A σ = 1.0 represents a membrane impermeable to protein; σ = 0 represents a membrane that is completely permeable to protein. In skin, the normal σ of albumin is reported to be 0.85–0.99.25,59 Increased capillary permeability to protein causes a reduced σ, an effective reduction in the reabsorptive oncotic gradient across the capillary wall and a resulting increase in net fluid filtration. Lymph sampled from burned skin has shown elevated protein concentrations consistent with the large and sustained increases in capillary permeability,15,56,59 whereas a transient and smaller increase in capillary permeability occurs over 8–12 hours following injury in other soft tissue not directly burned.56 Pitt et al.47 estimated the σ for skin from dog hindpaw using a lymph washdown technique and reported a normal σ of 0.87 for albumin and a reduction to 0.45 after scald injury.
Plasma colloid osmotic pressure (πp)
The normal plasma protein concentration of 6–8 g/dL, and its associated πp of 25–30 mmHg, produces a significant transcapillary reabsorptive force counterbalancing other Starling forces that favor filtration.14,25 Plasma colloid osmotic pressure decreases in non-resuscitated burn injured animals as protein-rich fluid extravasates into burn wounds, and a significant volume of protein-poor transcapillary reabsorption comes from non-burned tissue, such as skeletal muscle.14,51,52,60 Plasma is further diluted and πp is further reduced after crystalloid resuscitation. Zetterstrom and Arturson found that πp was reduced to half of the normal values in burn patients; πp can decrease so rapidly with resuscitation that the transcapillary colloid osmotic pressure gradient (πp – πif) will approach zero or even reverse to favor filtration and edema.51,52 Although it is likely that some hypoproteinemia is inevitable after major burn injury, animal and clinical studies using early colloid resuscitation produce higher levels of πp than with crystalloid resuscitation alone.7,60,61 The degree of hypoproteinemia and reduced πp were reported to correlate with the total volume of crystalloid solutions.60 Initial therapy with colloid solution has always been advocated by some clinicians,7 but the majority wait 8–24 hours after injury, reasoning that some normalization of microvascular permeability in injured tissue must occur before colloid therapy is cost effective.8 In recent years there appears to be greater use of albumin therapy earlier for burn resuscitation, and there is new evidence for improved outcomes with albumin resuscitation of burns.62,63
Interstitial colloid osmotic pressure (πif)
The πif in skin is normally 10–15 mmHg or about one-half that of plasma.14,25 Experimental studies in animals using lymph as representative of interstitial fluid suggest that the colloid osmotic pressure in lymph from burned skin initially increases 4–8 mmHg after burn injury.56 However, more direct measurements of πif using wick sampling14,20,52,64 or tissue sampling techniques14,25 show only modest initial increases in πif of 1–4 mmHg in the early non-resuscitated phase of burn injury. With resuscitation, πp falls and then πif decreases, as the protein concentration of capillary filtrate remains less than that of plasma despite an increased permeability. The osmotic reflection coefficient, σ, decreases with burns, but never equals zero, thus protein concentration in filtrate is less than in plasma even in burned skin.56 Compared to non-burned skin the πi remains significantly higher in the burn wound, supporting the view that sustained increases in protein permeability contribute to the persistence of burn edema.14,25,50 However, compared with the large changes in Pc and particularly Pif, increased capillary protein permeability is not the predominant mechanism for the early rapid rate of edema formation in injured skin.48
Non-burned tissue
Generalized edema in soft tissues not directly injured is another characteristic of large cutaneous burns. Brouhard et al.65 reported increased water content in non-burned skin even after a 10% burn, with the peak edema occurring 12 hours post burn. Arturson reported an increased transcapillary fluid flux (lymph flow) from non-burned tissue and a transient increase in permeability, as measured by an increase in the lymph concentration of plasma protein and macromolecular dextran infused as a tracer.15,21,59 Harms et al.56 extended these findings by measuring changes in lymph flow and protein transport in non-injured soft tissue for 3 days after injury. They found that skin and muscle permeability (flank lymph from sheep) were elevated for up to 12 hours post burn for molecules the size of albumin and immunoglobulin G, but the microvascular permeability of the lung (lymph for caudal mediastinal node) showed no increase. Maximum increased lymph flow and tissue water content were observed to correlate with the severe hypoproteinemia that occurred during the early resuscitation period of a 40% burn injury in sheep.8,61 The sustained increase in water content and the elevated lymph flow of the non-burned tissue after the return of normal permeability is likely the result of the sustained hypoproteinemia.51,56,59,60
Demling and colleagues66 suggested that the edema could be partially attributed to alterations in the interstitial structure. They suggested that interstitial protein washout increases the compliance of the interstitial space and that water transport and hydraulic conductivity across the entire blood–tissue–lymph barrier increased with hypoproteinemia. Several clinical and animal studies have established that maintaining higher levels of total plasma protein concentration can ameliorate the overall net fluid retention and edema.7,67 Non-burn edema can also be moderated by infusion of non-protein colloids such as dextran, if the colloid osmotic gradient is increased above normal.8,61 However, it is not known whether either the correction of hypoproteinemia or the use of either albumin or dextran leads to improved clinical outcome. It has been reported that the use of colloids has no beneficial effect on edema in the burn wound.50,61 Use of hypertonic saline formulations as initial fluid therapies for burn shock can greatly reduce initial volume requirements and net fluid volume (infused in minus urine out).68,69 However, a rebound of fluid requirements and net fluid can occur after early use of hypertonics and colloids.61,68 Retrospective analyses of patients correlating early albumin use with fluid requirements show significant volume sparing during the first post-burn day, but after 48 hours the effect is less apparent.70
Altered cellular membranes and cellular edema
In addition to a loss of capillary endothelial integrity, thermal injury also causes change in the cellular membrane. In skeletal muscle cellular transmembrane potentials decrease away from the site of injury.10 It would be expected that the directly injured cell would have a damaged cell membrane, increasing sodium and potassium fluxes and resulting in cell swelling. However, this process also occurs in cells that are not directly heat-injured. Micropuncture techniques have demonstrated partial depolarization in the normal skeletal muscle membrane potential of −90 mV to levels of −70 to −80 mV; cell death occurs at −60 mV. The decrease in membrane potentials is associated with an increase in intracellular water and sodium.71–73 Similar alterations in skeletal membrane functions and cellular edema have been reported in hemorrhagic shock71,73 and in cardiac, liver, and endothelial cells.74–76 Action potentials become dampened or non-existent, with likely delays in signal propagation in nerves, brain, skeletal muscle, heart, diaphragm, and gastrointestinal organs. Encephalopathy, muscle weakness, impaired cardiac contractility and gut dysfunction are associated with major burn injury, and may be due in part to reduced membrane potentials. Early investigators of this phenomenon postulated that a decrease in ATP levels or ATPase activity was the mechanism for membrane depolarization. However, more recent research suggests that it may result from an increased sodium conductance in membranes, or that an increase in sodium–hydrogen antiport activity is the primary mechanism.72,75 Resuscitation of hemorrhage rapidly restores depolarized membrane potentials to normal, but resuscitation of burn injury only partially restores the membrane potential and intracellular sodium concentrations to normal levels, demonstrating that hypovolemia alone is not totally responsible for the cellular swelling seen in burn shock.77 A circulating shock factor(s) is likely to be responsible for the membrane depolarization.78–80 When plasma from a burn-injured animal is superfused to an isolated muscle preparation, membrane depolarization occurs. Further, the depolarization can be reversed by changing the superfusion to normal plasma or saline.77 Surprisingly, the molecular characterization of such circulating factors have not been elucidated, suggesting that it has a complex and perhaps dynamic structure. Data suggest a large molecular weight, > 80 kDa.81 Membrane depolarization may be caused by different factors in different states of shock. Very little is known about the time course of the changes in membrane potential in clinical burns. More importantly, we do not know the extent to which the altered membrane potentials affect total volume requirements and organ function in burn injury, or even shock in general.
Inflammatory mediators of burn injury
Histamine
Histamine is a key mediator responsible for the early phase of increased microvascular permeability seen immediately after burn. Histamine causes large endothelial gaps to transiently form as a result of the contraction of venular endothelial cells.28 Histamine is released from mast cells in thermally injured skin; however, the increase in histamine levels and its actions are only transient. Histamine also can cause the rise in capillary pressure (Pc) by arteriolar dilation and venular contraction. Statistically significant reductions in localized edema have been achieved with histamine blockers and mast cell stabilizers when tested in animal models.28 Friedl et al.30 demonstrated that the pathogenesis of burn edema in the skin of rats appears to be related to the interaction of histamine with xanthine oxidase and oxygen radicals. Histamine and its metabolic derivatives increased the catalytic activity of xanthine oxidase (but not xanthine dehydrogenase) in rat plasma and in rat pulmonary artery endothelial cells. In thermally injured rats, levels of plasma histamine and xanthine oxidase rose in parallel, in association with the increase in uric acid. Burn edema was greatly attenuated by treating rats with the mast cell stabilizer cromolyn, complement depletion, or the H2 receptor antagonist cimetidine, but was unaffected by neutrophil depletion.29,34,82 Despite encouraging results in animals, beneficial antihistamine treatment of human burn injury has not been demonstrated, although antihistamines are administered to reduce the risk of gastric ulcers.
Prostaglandins
Prostaglandins are potent vasoactive autacoids synthesized from the arachidonic acid released from burned tissue and inflammatory cells, and contribute to the inflammatory response of burn injury.36,83 Activated macrophages and neutrophils infiltrate the wound and release prostaglandin as well as thromboxanes, leukotrienes and interleukin (IL)-1. These wound mediators have both local and systemic effects. Prostaglandin E2 (PGE2) and leukotrienes LB4 and LD4 increase microvascular permeability both directly and indirectly.84 Prostacyclin (PGI2) is produced in burn injury and is also a vasodilator, but also may cause direct increases in capillary permeability. PGE2 appears to be one of the more potent inflammatory prostaglandins, causing postburn vasodilation and increased microvascular surface area in wounds, which when coupled with the increased microvascular permeability amplifies edema formation.85,86
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


