Introduction
Shock is an abnormal physiologic state in which tissue perfusion is insufficient for oxygen and nutrient delivery and cellular waste removal. Extensive cutaneous thermal injury (≥20% of the total body surface area [TBSA]) invariably results in severe derangements of cardiovascular function and end-organ perfusion known as burn shock. Whereas burn patients may sustain episodes of septic or hemorrhagic shock at other times during a long intensive care unit (ICU) course, the term burn shock refers only to the first 24 to 48 hours postburn. Burn shock is a dynamically changing problem. Understanding the typical trajectory as a patient moves through burn shock and changes in response to fluid resuscitation is essential for successful patient care. Furthermore, failure of resuscitation historically occurred in about 13% of patients with burn shock, sometimes as a result of overresuscitation leading to lethal abdominal compartment syndrome (ACS). Thus burn shock is a high-risk problem for the burn team.
Burn shock is the result of three main processes: (1) hypovolemia caused by loss of fluid from the intravascular space into the interstitial space (and, to a lesser extent, externally), (2) increased systemic and pulmonary vascular resistance, and (3) decreased intrinsic myocardial contractility caused by circulating mediators. This complex process of microcirculatory dysfunction and circulatory failure can be mitigated by fluid resuscitation but, to date, not repaired by medical therapy.
This chapter examines our current understanding of the pathophysiology of burn shock, focusing in turn on the systemic and pulmonary circulation, the microvasculature, circulating mediators, and end-organ effects. Intracellular pathways are not presented.
The systemic circulation
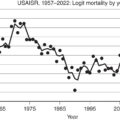
The classic study by Pruitt et al. provides a useful frame of reference for the dynamic changes in hemodynamics and volume status that occur during burn shock ( Fig. 7.1 ). There is a massive and sustained release of catecholamines and other vasoconstrictors , and an immediate depression of cardiac output (CO) after thermal injury, before any detectable reduction in plasma volume. The rapidity of this response may result from impaired neurotransmission of cardiac signaling and increased afterload due to vasoconstriction. Soon after injury, a developing hypovolemia and reduced venous return undeniably contribute to the reduced CO.
Hemodynamic changes during resuscitation of patients with burn shock.
(A) Changes in plasma and blood volume. (B) Changes in cardiac output and systemic vascular resistance (peripheral resistance).
(From Pruitt Jr BA, Mason Jr AD, Moncrief JA. Hemodynamic changes in the early postburn patient: The influence of fluid administration and of a vasodilator [hydralazine]. J Trauma . 1971;11[1]:36-46.)
The adrenergic response causes tachycardia and tends to maintain the blood pressure, even while blood volume, CO, and end-organ perfusion fall. Thus a normal blood pressure does not mean that the patient is not in shock.
Hypovolemia
Burn shock predominantly features hypovolemia and is characterized by hemodynamic changes similar to those that occur after hemorrhage. These include decreased plasma volume, CO, and urine output; in addition, increased systemic vascular resistance (SVR) occurs with resultant reduced blood flow to vital organs. , , Whereas in hemorrhage there is a fall in hematocrit, in burn shock hematocrit often rises due to loss of plasma rather than blood. This is particularly common when fluid therapy is inadequate.
Large volumes of resuscitation solutions are required to maintain intravascular volume during the first 24 to 48 hours after an extensive burn, but resuscitation is complicated by the occurrence of edema in both burned and—in larger burns (typically >25% of TBSA)—unburned soft tissue. Excessive fluid resuscitation, by increasing pressures across the microvasculature and by damaging the endothelial glycocalyx, drives edema formation and can lead to complications such as compartment syndrome. The process of edema formation is further discussed later in the chapter.
Systemic and pulmonary vascular resistance
Michie et al. measured CO and SVR in anesthetized dogs resuscitated after burn injury. They found that CO fell shortly after injury and then returned toward normal; however, reduced CO did not parallel the blood volume deficit. They concluded that the depression of CO resulted not only from decreased blood volume and venous return but also from an increased SVR and from the presence of a circulating myocardial depressant substance.
Increased afterload after burn injury is the result of release of catecholamines, vasopressin, angiotensin II, and neuropeptide Y. , These agents cause contraction of the arteriolar smooth muscle, which is systemically manifested by increased afterload and SVR. The increased SVR after burn injury is also, in part, the result of increased blood viscosity secondary to hemoconcentration. Several investigators have studied the use of vasodilators to address the increased SVR and thereby to increase CO. For example, Pruitt et al. found that hydralazine could be used for this purpose, with the caveat that hypovolemia should first be corrected. Another option would be dobutamine, with the same caveat about hypovolemia.
There are similarities and differences in the responses seen in the pulmonary circulation in comparison to the systemic circulation. In large burns there is a pronounced increase in pulmonary vascular resistance (PVR), but this PVR increase is sustained longer (resolving 42 hours postburn) than the SVR increase (resolving 18–24 hours postburn). The effects of burn shock on pulmonary function are further discussed in “End-Organ Effects.”
Myocardial depression
Postburn depression of intrinsic myocardial contractility by circulating factors was first suggested by Baxter, who performed cross-circulation studies in dogs while assigned to the US Army Surgical Research Unit. Such myocardial dysfunction was subsequently demonstrated in numerous isolated heart studies (Langendorff preparation). Aggressive fluid resuscitation, alone, is not sufficient to correct these left ventricular contractile and compliance defects.
No one myocardial depressant factor has been conclusively identified; rather, several pathophysiologic processes have been implicated. These include the proinflammatory cytokines (tumor necrosis factor α [TNF-α], interleukin 1β [IL-1β], IL-6); high-mobility group box 1 (HMGB-1), macrophage migration inhibitory factor, heat shock proteins, apoptosis, caspases, and oxygen-derived free radicals. , Clinically, myocardial dysfunction may not be evident because in intact subjects the heart is exposed to circulating catecholamines and is influenced by cardiac sympathetic nerve activity. Thus Goodwin et al. demonstrated hyperdynamic left ventricular activity by echocardiography in their trial of resuscitation fluids. On the other hand, Howard et al. demonstrated both systolic and diastolic dysfunction in burn-injured children during the first few weeks postinjury. Older patients (and older animals) are particularly likely to respond to burn shock with a hypodynamic myocardium.
Edema formation
During burn shock, hypovolemia is paralleled by edema formation. Edema develops when the rate at which fluid is filtered out of the capillaries exceeds the flow in the lymph vessels. Edema formation often follows a biphasic pattern. An immediate and rapid increase in the water content of burned tissue is seen in the first hour postburn. , A second and more gradual increase in fluid content in both burned skin and unburned soft tissue occurs during the first 12 to 24 hours postburn.
The amount of edema formation in burned skin depends on the depth and extent of injury, , on whether fluid resuscitation is provided, and on the type and volume of fluid administered. Fluid resuscitation elevates blood flow and capillary pressure, thereby contributing to further fluid extravasation. Without sustained IV replacement of intravascular fluid losses, edema formation is somewhat self-limited, as tissue blood flow and capillary pressure decrease.
Early edema formation in thermally injured skin is characterized by an extremely rapid onset. Tissue water content can double within the first hour after burn. Leape found a 70% to 80% increase in water content in a full-thickness burn wound 30 minutes after burn injury, with 90% of this change occurring in the first 5 minutes. , , There was only a modest increase in burn wound water content after the first hour in unresuscitated animals. In resuscitated animals or animals with small wounds, adequate tissue perfusion continues to feed the edema for several hours. Demling et al. used dichromatic absorptiometry to measure edema development during the first week after an experimental partial-thickness burn injury on one hind limb in sheep. Although edema formation was rapid, with more than 50% occurring in the first hour, maximum water content was not present until 12 to 24 hours after burn injury. The mass of the burned tissue was significantly less than that of the remainder of the body. As such, most fluid shifts likely occur from the blood into the unburned tissue due to the actions of circulating inflammatory mediators on the endothelium.
Microcirculatory fluid exchange
Fluid losses and edema formation during burn shock are the result of a constellation of changes in pressure and permeability across the microvasculature. Under physiologic steady-state conditions, blood pressure in capillaries causes filtration of fluid into the interstitial space. The bulk of this filtrate is removed from the interstitial space by lymphatic drainage. Fluid transport across the microcirculatory wall has been described by the classic Starling equation:
Here, J v is the flux (flow rate) of fluid that crosses the microvasculature barrier. K f is the capillary filtration coefficient, which is the product of the surface area and hydraulic conductivity (water permeability) of the capillary wall. Additionally, P c is the capillary hydrostatic pressure, P if is the interstitial fluid hydrostatic pressure, π p is the colloid osmotic pressure of plasma, π if is the colloid osmotic pressure of interstitial fluid, and σ is the osmotic reflection coefficient. Edema occurs when the lymphatic drainage rate ( J L ) does not keep pace with the increased J v . Fig. 7.2 shows the key structures and microvascular forces of the classic Starling equation. Thermal injury is unique compared to other edematous disorders because only in burns do all of the Starling equation variables change in the direction that increases fluid filtration. We will review each term of the classic Starling equation and its role in burn edema before addressing possible implications of a Starling equation modified to take into account the endothelial glycocalyx.
The classic Starling equation and microvascular forces.
Capillary filtration coefficient
K f is a function of both the capillary surface area and its water permeability. Thus local vasodilation and microvascular recruitment contribute to the increased K f postburn. Furthermore, burn injury causes direct and indirect (mediator-driven) changes in the water permeability of the blood–tissue barrier of the capillaries and venules. Arturson and Mellander showed that, in the scalded hindlimb of dogs, K f immediately increased two to three times, suggesting that the water permeability of the capillary wall increased. Measuring K f and the rate of edema formation ( J v ) allowed them to determine that a transcapillary pressure gradient of 100 to 250 mm Hg would be required to explain the extremely rapid edema formation that occurred in the first 10 minutes after a scald injury. Subsequent studies described herein show that these large increases in filtration forces also result from an increase in P c and a decrease in P if .
Capillary pressure
In most forms of shock, arteriolar vasoconstriction results in transfer of less arterial pressure to the capillaries; capillary and venous pressures decrease. However, in studies using the vascular occlusion technique in the scalded hindlimb of dogs, P c doubled from approximately 25 mmHg to approximately 50 mmHg during the first 30 minutes after burn injury and slowly returned to baseline over 3 hours. This points to early arteriolar dilation and venular constriction.
Interstitial hydrostatic pressure
Burned tissue has been demonstrated to have a significantly decreased interstitial hydrostatic pressure. Using micropipettes and a tissue oncometer, Lund et al. reported that dermal P if was rapidly reduced from its normal value of −1 mmHg to less than −100 mmHg in isolated nonperfused samples of skin. This large negative interstitial hydrostatic pressure constitutes a powerful suction force or imbibition pressure promoting microvascular fluid filtration and sustained burn wound edema formation. In vivo measurements show a temporary reduction of −20 to −30 mmHg; the less negative P if in vivo is due to the continued tissue perfusion and fluid extravasation that relieves the imbibition pressure. Kinsky et al. reported a continued negative P if , providing a partial explanation for sustained edema formation during the first 4 hours postburn.
The large decrease in P if is caused, at least in part, by the release of cellular tension exerted on the collagen and microfibril networks in the connective tissue via the collagen-binding β 1 integrins. This tends to expand the interstitial space and induces the imbibition pressure. The integrins are transmembrane adhesion receptors that mediate cell–cell and cell–matrix adhesion, thereby allowing the glycosaminoglycan ground substance, which is normally underhydrated, to expand and take up fluid. McGee et al. confirmed this hydration potential with T2-weighted MRI and noted that it is reversible by application of negative pressure. This supports the mechanism of interfascial rather than colloid-osmotic fluid transfer as a mechanism for burn edema.
Osmotic reflection coefficient
The osmotic reflection coefficient is an index of microvascular permeability to protein. A value of σ = 1.0 represents a barrier impermeable to protein but permeable to water, and σ = 0 represents a barrier that is completely permeable to both protein and water. The reflection coefficient is traditionally attributed to the endothelial cellular junctions, but may well be primarily determined by the glycocalyx. In normal skin, the σ of albumin is reported to be 0.85 to 0.99. , Thermal injury causes an increase in capillary permeability to protein, resulting in a reduced σ, and a resulting increase in net fluid filtration. Lymph sampled from burned skin has shown elevated protein concentrations consistent with the large and sustained increases in capillary permeability, , , whereas a transient and smaller increase in microvascular permeability occurs following injury in unburned soft tissue. Pitt et al. estimated the σ for skin from dog hindpaw using a lymph wash-down technique and reported a normal σ of 0.87 for albumin and a reduction to 0.45 after scald injury.
Plasma colloid osmotic pressure
The normal plasma protein concentration of 6 to 8 g/dL, and its associated π p of 25 to 30 mmHg, would produce a significant transcapillary absorptive force counterbalancing the other Starling forces that favor filtration. , Hypoproteinemia develops, and π p decreases, in unresuscitated burned animals as protein-rich fluid extravasates into burn wounds; protein-poor interstitial fluid enters the circulation from transvascular reabsorption and from unburned tissue lymph. , Plasma is further diluted, and π p is reduced after crystalloid resuscitation. Addressing this developing hypoproteinemia is one rationale for colloid-based resuscitation.
Interstitial colloid osmotic pressure
The π if in skin is generated by proteins in the interstitial space, and is normally 10 to 15 mmHg or about one-half that of plasma. , Experimental studies in animals using lymph as representative of interstitial fluid suggest that the π if in lymph from burned skin initially increases 4 to 8 mmHg postburn. With crystalloid resuscitation, both π p and π if later decrease. The osmotic reflection coefficient (σ) decreases but never equals zero; thus protein concentration in capillary filtrate is always less than in plasma, even in burned skin. Compared to unburned skin, the π i f remains significantly higher in the burn wound, supporting the view that sustained increases in protein permeability contribute to the persistence of burn edema. , , However, compared with the large changes in P c and particularly P if , increased microvascular protein permeability is not the predominant mechanism for the early, rapid rate of edema formation in injured skin.
Endothelial dysfunction and the glycocalyx
The glycocalyx is a glycoprotein and polysaccharide layer on the luminal side of endothelial cells. It maintains the barrier between the endothelium and plasma by reducing the osmotic gradient and thereby reducing filtration. Based on studies of the glycocalyx, the classic Starling equation has been challenged, and a revised equation has been proposed ( Fig. 7.3 ) :
The revised Starling equation, taking into account the role of the glycocalyx.
In the revised Starling equation, the fluid flux across the blood–tissue barrier is driven primarily by differences in hydrostatic pressures ( P c − P if ) and by a colloid osmotic gradient between the fluid in the glycocalyx and just below the glycocalyx (π p −π g ).
Studies by Kozar et al. have demonstrated that crystalloid resuscitation of hemorrhagic shock in rats causes a loss of glycocalyx that can be largely prevented by plasma resuscitation. Shupp and others have identified a burn size–dependent relationship between thermal injury and the release of syndecan 1, indicating that glycocalyx breakdown occurs postburn. Similar to Kozar, they also found that plasma as opposed to crystalloid resuscitation tended to protect the glycocalyx, although the components of plasma responsible for this effect remain to be determined.
Edema in unburned tissue
Generalized edema in soft tissues not directly injured is another characteristic of large cutaneous burns. Arturson reported increased transvascular fluid flux (lymph flow) from unburned tissue and a transient increase in permeability as measured by an increase in the lymph concentration of plasma protein and macromolecular dextran infused as a tracer. , Harms et al. extended these findings by measuring changes in lymph flow and protein transport in uninjured soft tissue for 3 days after injury. They found that skin and muscle permeability (flank lymph from sheep) was elevated for up to 12 hours postburn for molecules the size of albumin and immunoglobulin G, but the microvascular permeability of the lung (lymph from caudal mediastinal node) showed no increase. Maximum increased lymph flow and tissue water content were observed to correlate with the severe hypoproteinemia that occurred during the early resuscitation period of a 40% burn injury in sheep.
The mechanisms by which burn injury induces microvascular hyperpermeability remote from the injury have been the subject of extensive study utilizing plasma from burned animals transfused into unburned recipients by Kremer et al. These transfusions cause endothelial activation, albumin leakage, leukocyte adhesion, and rolling via undefined transfused circulating factor(s).
Cellular membrane depolarization
In addition to a loss of microvascular barrier integrity, thermal injury also causes changes in the cell membrane. In skeletal muscle, cell transmembrane potentials decrease at sites distant from the injury. These changes are well described in hemorrhagic shock in skeletal muscle, cardiac muscle, hepatocytes, and endothelial cells. Micropuncture techniques in hemorrhaged animals have demonstrated partial depolarization in the skeletal muscle membrane potential of −90 mV to levels of −70 to −80 mV; cell death occurs at −60 mV. These decreases in membrane potentials are associated with increases in intracellular water and sodium. Action potentials become dampened or nonexistent, with likely delays in signal propagation in nerves, brain, skeletal muscle, heart, diaphragm, and gastrointestinal organs. Encephalopathy, muscle weakness, impaired cardiac contractility, and gut dysfunction are associated with major burn injury and may be due in part to reduced membrane potentials.
Early investigators of this phenomenon postulated that a decrease in adenosine triphosphate (ATP) levels or ATPase activity was the mechanism for membrane depolarization. However, more recent research suggests that it may result from an increased sodium conductance in membranes or that an increase in sodium–hydrogen antiporter activity is the primary mechanism. , Resuscitation of hemorrhage rapidly restores membrane potentials to normal, but resuscitation of burn injury only partially restores membrane potentials and intracellular sodium concentrations to normal, demonstrating that hypovolemia alone is not solely responsible for the cellular swelling seen in burn shock. Circulating shock factor(s) has been postulated to contribute to membrane depolarization. When plasma from a burn-injured animal is superfused to an isolated muscle preparation, membrane depolarization occurs. Furthermore, the depolarization can be reversed by changing the superfusate to plasma or saline.
Inflammatory mediators
Substantial research has focused on identifying and defining the mechanisms and effects of the many inflammatory mediators produced and released after burn injury. These mediators play complex roles in the pathogenesis of edema and the cardiovascular abnormalities of burn injury. For example, mediators alter vascular permeability and transvascular fluid flux, either directly or indirectly. Unfortunately, most strategies directed at mediator blockade have had no clinical impact on the care of patients with major burns.
Histamine
Histamine is a key mediator of very early increases in microvascular permeability following thermal injury. Histamine is released from mast cells in thermally injured skin; however, this increase and its effects are only transient. Histamine causes large endothelial gaps to transiently form as a result of the contraction of venular endothelial cells. Histamine also contributes to the rise in capillary pressure ( P c ) by arteriolar dilation and venular contraction. Reductions in localized edema have been achieved with histamine blockers and mast cell stabilizers when tested in animal models. Friedl et al. demonstrated that the pathogenesis of burn edema in the skin of rats appears to be related to the interaction of histamine with xanthine oxidase and oxygen radicals. Histamine and its metabolic derivatives increased the catalytic activity of xanthine oxidase in rat plasma and in rat pulmonary artery endothelial cells. In thermally injured rats, levels of plasma histamine and xanthine oxidase rose in parallel, in association with the increase in uric acid. Burn edema was greatly attenuated by treating rats with the mast cell stabilizer cromolyn, complement depletion, or the H 2 -receptor antagonist cimetidine but was unaffected by neutrophil depletion. Despite encouraging results in animals, successful antihistamine treatment of human burn injury has not been demonstrated.
Kinins
Bradykinin is a local mediator of inflammation that increases venular permeability. It is likely that bradykinin production is increased after burn injury, but its detection in blood or lymph can be difficult because of the simultaneous increase in kininase activity and the rapid inactivation of free kinins. The generalized inflammatory response after burn injury favors the release of bradykinin. Pretreatment of burned animals with aprotinin, a general protease inhibitor, should have decreased the release of free kinin, but no effect on edema was noted. On the other hand, pretreatment with a specific bradykinin receptor antagonist was reported to reduce edema in burn wounds in rabbits. Tao et al. demonstrated that blocking neurokinin 1 decreased the vascular permeability tissue around the wound and remotely in the jejunum of burned rats, and that treated rats recovered more quickly than controls.
Damage-associated molecular patterns and the cytokine storm
Damage-associated molecular patterns (DAMPs) released from dead, dying, or injured cells are now recognized as key initiators of early postburn inflammation. These DAMPs include HMGB-1, heat shock proteins, ATP, DNA, and components of the extracellular matrix and cell membrane. DAMPs interact with pathogen recognition receptors (PRRs) on macrophages, neutrophils, and endothelial cells. Such PRRs include the Toll-like receptors and advanced glycosylation end product–specific receptors. This leads to increased production of an array of cytokines with resulting end-organ dysfunction (and later immunosuppression). For example, in a prospective observational study of burn patients, cytokines IL-6, IL-8, IL-10, and monocyte chemoattractant protein 1 (MCP-1) were associated with Sequential Organ Failure Assessment (SOFA) scores on day 5 and 1 month postburn. There was a correlation between levels of IL-6, IL-8, and IL-10 on days 1 and 2, and 28-day mortality.
There are several potential therapeutic options that target the DAMP-mediated cytokine storm. Topical p38 mitogen-activated protein kinase inhibitor and tranexamic acid (TXA) significantly attenuated mitochondrial DNA (mtDNA) release and reduced lung inflammation in a murine model. The mechanism of TXA action in this model is uncertain but may involve suppression of nuclear factor κB pathway signaling. In a rat study, 17β-estradiol was shown to exert a plethora of protective effects on myocardial function, mediated by reduced production of mtDNA, mitochondrial reactive oxygen species (ROS), and cytochrome C.
Prostaglandins
Prostaglandins are potent vasoactive autocoids synthesized from the arachidonic acid released from burned tissue and inflammatory cells, and they contribute to the inflammatory response following burn injury. , Activated macrophages and neutrophils infiltrate the wound and release prostaglandins as well as thromboxanes, leukotrienes, and IL-1. These wound mediators have both local and systemic effects. Prostaglandin E 2 (PGE 2 ) and leukotrienes LB 4 and LD 4 increase microvascular permeability both directly and indirectly. PGE 2 appears to be one of the more potent inflammatory prostaglandins, causing postburn vasodilation and increased microvascular surface area in wounds that, when coupled with the increased microvascular permeability, amplifies edema formation. , Prostacyclin (PGI 2 ) is a vasodilator and may cause increases in capillary permeability.
Thromboxane
Thromboxane A 2 (TXA 2 ) and its metabolite, thromboxane B 2 (TXB 2 ), are produced locally in burn wounds by platelets. Vasoconstrictor thromboxanes may be less important in edema formation; however, by reducing blood flow they can contribute to a growing zone of ischemia within the burn wound and may be responsible in part for the conversion of a partial-thickness wound to a deeper, full-thickness wound. The serum level of TXA 2 and TXA 2 /PGI 2 ratio are significantly increased in burn patients. Heggers et al. showed that TXB 2 is released at the burn wound and is associated with local tissue ischemia, while thromboxane inhibitors prevented the progressive dermal ischemia associated with thermal injury and thromboxane release. , The TXA 2 synthesis inhibitor anisodamine also showed beneficial microcirculatory effects. LaLonde et al. showed that topically applied ibuprofen (which inhibits the synthesis of prostaglandins and thromboxanes) reduced both local edema and prostanoid production in burned tissue without altering systemic production. On the other hand, systemic administration of ibuprofen did not modify early edema, but it did attenuate the postburn vasoconstriction that impaired adequate oxygen delivery to tissue in burned sheep. Although cyclooxygenase inhibitors have been used after burn injury, they have not entered into routine clinical use.
Serotonin
Serotonin is released early after burn injury. This agent is a smooth muscle constrictor of large blood vessels. Antiserotonin agents such as ketanserin have been found to reduce peripheral vascular resistance after burn injury but not to reduce edema. On the other hand, pretreatment with methysergide, a serotonin antagonist, reduces hyperemia in the burn wounds of rabbits and reduces burn edema. Ferrara et al. found a dose-dependent reduction in burn edema when methysergide was given to dogs prior to burn injury, but claimed that this was not attributable to blunting of the regional vasodilation response. Zhang et al. reported a reduction in skin blood flow after methysergide administration to burned rabbits. Ketanserin, a 5-HT 2a (serotonin receptor) antagonist, reduced plasma extravasation and leukocyte–endothelial interactions after burn plasma transfer. These findings were further confirmed with cinanserin and methysergide. ,
Catecholamines
Epinephrine and norepinephrine are increased several fold after burn injury. , In addition to the central nervous system and adrenal glands, macrophages are now recognized as an important source of catecholamines following challenges such as hemorrhagic shock and resuscitation or lipopolysaccharide-induced acute lung injury. Via α 1 -receptor activation, these agents cause arteriolar vasoconstriction, which tends to reduce capillary pressure. Via β-agonist activity, they may also inhibit the increased microvascular permeability induced by histamine and bradykinin. These potentially beneficial effects of catecholamines may not be operative in injured tissue because of the effects of other mediators. Their systemic hemodynamic effects were previously discussed.
Angiotensin II and vasopressin
During burn shock, sympathetic tone is high and volume receptors sense hypovolemia, both of which elevate angiotensin II and vasopressin to supranormal levels in the blood. Both are potent vasoconstrictors of terminal arterioles with less effect on the venules. Angiotensin II may be responsible for selective gut and mucosal ischemia. In severely burned patients, angiotensin II levels were 2 to 8 times normal on the first 1 to 5 days postburn, with peak levels occurring on day 3. Vasopressin levels were 50 times normal upon admission and declined toward normal over the first 5 days postburn. Along with catecholamines, vasopressin may be largely responsible for increased SVR. Sun et al. used vasopressin receptor antagonists to improve hemodynamics and survival time in rats with burn shock, whereas vasopressin infusion exacerbated burn shock.
Reactive oxygen species
ROS, also known as oxygen-derived free radicals, play an important role in all forms of shock. These short-lived elements are highly unstable reactive metabolites of oxygen; each one has an unpaired electron, making all of them strong oxidizing agents. Superoxide anion (O 2 − ), hydrogen peroxide (H 2 O 2 ), and hydroxyl ion are produced and released by activated neutrophils after any inflammatory reaction or reperfusion of ischemic tissue. Increased lipid peroxidation found in circulating red blood cells and in biopsied tissue provides evidence that these agents are formed after burn injury.
Antioxidants, namely agents that either bind directly to the oxygen radicals (scavengers) or cause their further metabolism, have been evaluated in several models. Catalase removes H 2 O 2 and superoxide dismutase (SOD), lessens radical O 2 − , and is reported to reduce plasma loss after burn injury in dogs and rats.
Xanthine oxidase catalyzes the production of ROS. The plasma of thermally injured rats showed dramatic increases in levels of xanthine oxidase activity, with peak values appearing as early as 15 minutes after thermal injury. Excision of the burned skin immediately after the injury significantly diminished the increase in plasma xanthine oxidase activity. , The skin permeability changes were attenuated by treating the animals with antioxidants (catalase, SOD, dimethyl sulfoxide, dimethylthiourea) or an iron chelator (deferoxamine), thereby supporting the role of oxygen radicals in the development of vascular injury as defined by increased vascular permeability. Allopurinol, a xanthine oxidase inhibitor, markedly reduced both burn lymph flow and levels of circulating lipid peroxides and further prevented all pulmonary lipid peroxidation and inflammation. This suggests that the release of oxidants from burned tissue was in part responsible for local burn edema as well as for systemic inflammation and oxidant release. The failure of neutrophil depletion to protect against the vascular permeability changes and the protective effects of the xanthine oxidase inhibitors (allopurinol and lodoxamide tromethamine) suggests that plasma xanthine oxidase is the more likely source of the oxygen radicals involved in the formation of burn edema. These oxygen radicals can increase vascular permeability by damaging microvascular endothelial cells. ,
High doses of the antioxidant vitamin C have been found to be efficacious in reducing fluid needs in animal models when administered postburn. High-dose vitamin C (66 mg/kg/hour) was shown to reduce volume requirements in one clinical trial ; the burn community awaits more definitive trials.
Nitric oxide
Nitric oxide (NO) is an important inhibitor of vascular smooth muscle tone. Following injury, activation of endothelial nitric oxide synthase and, more importantly, synthesis of inducible nitric oxide synthase (iNOS) lead to increased NO production and thereby vasodilation. Such increased NO production may be a primary driver of vasoplegia (a low CO, low SVR shock state in certain clinical settings such as septic shock and after cardiopulmonary bypass). Vasoplegia in burn patients has been described in case reports. , Methylene blue, which inhibits guanylate cyclase and thus the vasodilatory activity of NO, has been used to treat patients with this syndrome. Since the effects of iNOS take time to unfold, it is not surprising that vasoplegia is not clinically evident during the high-SVR early phase of burn shock.
NO generated simultaneously with the superoxide anion can lead to the formation of peroxynitrite. The presence of nitrotyrosine in burned skin in the first few hours after injury suggests that peroxynitrite may play a deleterious role in burn edema. But blockade of NOS did not reduce burn edema, whereas treatment with the NO precursor arginine did. Thus NO may be important for maintaining perfusion and limiting the zone of stasis in burn skin, and the value of NO reduction in burns remains controversial.
Platelet aggregation factor
Platelet aggregation (or activating) factor (PAF) can increase capillary permeability and is released after burn injury. , Ono et al. showed in scald-injured rabbits that the PAF antagonist TCV-309, infused soon postburn, blocked edema formation in the wound and decreased PAF levels in the damaged tissue. SOD content in the group treated with TCV-309 was significantly higher than that of the control group, implying less SOD consumption (and lower superoxide levels) in the treatment group.
Other mediators and pathways
-
Hydrogen sulfide (H 2 S) is a small molecule (gasotransmitter) capable of both inducing and inhibiting inflammation. The potential role of H 2 S in burn shock remains to be defined. ,
-
Cholinergic Antiinflammatory Pathway . Hu et al. used a 50% TBSA burn-injured dog model and reported that use of a nicotinic receptor antagonist with resuscitation increased survival, improved hemodynamics, increased plasma volume and urine output, and decreased TNF-α, IL-1, and lactic acid. In burn plasma transfer studies, Hernekamp et al. , demonstrated that the cholinergic antiinflammatory pathway, stimulated by CDP-choline or physostigmine, can attenuate albumin efflux and leukocyte adhesion.
-
Metalloproteinases (MMP). After burn-serum exposure, lung endothelial cells had increased matrix MMP activity and decreased tissue inhibitor of MMP (TIMP-2). These cells had increased monolayer permeability and damaged adhesion junction proteins and formation of actin stress fibers. This damage was inhibited effectively by exogenous TIMP-2.
-
Endothelin 1 (ET-1) is an important mediator of increased microvascular permeability in various tissue beds. In rats with burn injury, Jiang et al. found upregulation of hypoxia-inducible factor 1α and increased levels of ET-1 and its receptors. ET-1 induces degradation and translocation of vascular endothelial-cadherin and claudin-5 proteins, thus disrupting endothelial cell junction integrity and increasing permeability.
-
Coagulation Cascade. Burn shock is typically associated with a hypercoagulable state. It is reasonable to assume that at the microvascular level this phenomenon contributes to progressive burn-wound ischemia. Huzar et al. performed rapid thromboelastography on admission of patients with burns greater than 15% TBSA. They found that 21% were hypocoagulable, 17% were normal, and 62% were hypercoagulable. Hypocoagulable patients trended toward higher in-hospital mortality. Infrequently, burn patients are diagnosed with disseminated intravascular coagulation, a phenomenon that is also associated with increased mortality.
End-organ effects
Consistent with the ebb and flow model of the response to trauma, Pruitt succinctly summarized the end-organ effects of thermal injury as early hypofunction, followed by later hyperfunction; the nervous and immune systems displayed the opposite pattern. There are several organs particularly susceptible to early postburn dysfunction, mediated in part by decreased regional blood flow. The burn shock–induced decrease in CO (and increase in SVR) does not affect all organs equally. Asch et al. reported regional blood flow changes in burn shock in a canine model. Regional blood flow decreased to the kidneys, liver (hepatic artery), small intestine, and carcass. Fluid resuscitation sufficient to restore CO to baseline fully restored blood flow to the liver and carcass only. Other organ systems are susceptible to dysfunction despite adequate perfusion.
Central nervous system
Delirium is a vexingly common problem in extensively burned patients. In general ICU patients, delirium increases both in-hospital and postdischarge mortality. , Risk factors for delirium in burn patients include an American Society of Anesthesiologists score of 3 or more, TBSA greater than 10%, ICU admission, and need for surgery. Multiple factors contribute to delirium in these patients, some of which appear to be incited by burn shock. The systemic response to burn shock may disrupt the blood–brain barrier (BBB) and predispose to cerebral edema. One group placed epidural catheters into patients with burn size greater than 60% TBSA. They found average peak intracranial pressure (ICP) values of 31 ± 10 mmHg on day 2 postburn. Studies in anesthetized sheep subjected to a 70% TBSA scald showed that cerebral autoregulation is well maintained in the immediate postburn period. At the end of the 6-hour study, however, cerebrovascular resistance increased, cerebrovascular blood flow decreased, and ICP increased; there was increased brain water content at autopsy. In a rat model, there was a postburn increase in BBB permeability to labeled albumin. There was an increase in TNF-α, IL-1β, and ICAM-1 mRNA in the brain at 3 hours, prior to appearance of these molecules at 7 hours. There was increased expression of MMP-2 and MMP-9 in the brain, enzymes which degrade the BBB. Gatson et al. found that brain levels of TNF-α, IL-1β, and IL-6 were much higher than systemic levels. These data suggest that the brain participates in the inflammatory response to burn shock and that the BBB is vulnerable to injury.
Lungs
In contrast to the systemic circulation, pulmonary edema is unusual during burn shock, and typically does not become evident until after fluid resuscitation is complete. Possibly, sustained high levels of PVR (see earlier) play a protective role during burn shock. Pulmonary capillary wedge pressure (PCWP) is increased more than left atrial pressure after experimental burn injury due to postcapillary venular constriction. , By increasing PCWP, venular constriction may contribute to pulmonary edema. It is likely that some degree of left heart failure also contributes to the increased capillary pressure. However, hypoproteinemia may be the greatest contributing factor to postburn pulmonary edema. Analysis of lung lymph sampled in large animal models after 40% TBSA burn injury showed no evidence of increased microvascular permeability. Furthermore, lung lymph flow may increase considerably to counteract interstitial fluid accumulation. Consistent with these findings, clinical studies suggest that acute respiratory distress syndrome (ARDS) during burn shock is unusual; its occurrence (in the absence of inhalation injury or other direct lung insult) suggests a particularly florid response to a severe cutaneous burn.
It is critical to point out that patients with larger burns may require early endotracheal intubation even in the absence of inhalation injury or ARDS. In the study by Zak et al. of scald-injured children, those less than 2.8 years old and with burns greater than 19% TBSA were at risk of requiring early intubation. The authors speculated that edema of the smaller-diameter upper airways or reduced pulmonary compliance was a contributing factor. Pulmonary dysfunction associated with inhalation injury is discussed in a separate chapter.
Kidneys
Postburn renal failure rates declined dramatically because of standardized regimens of fluid resuscitation, but they remain surprisingly common and increase mortality. In a study of 744 patients admitted to the US Army Burn Center from recent conflicts in Iraq and Afghanistan, the prevalence of acute kidney injury (AKI) by RIFLE (risk, injury, failure, loss, end stage) criteria was 23.8% and by Acute Kidney Injury Network (AKIN) criteria was 29.9%. Age, TBSA, injury severity score, and AKIN were independent predictors of death by logistic regression. Most AKI in that study was diagnosed on the day of admission to the burn center (following aeromedical evacuation from overseas).
The importance of timely resuscitation in preserving renal function was suggested by Chrysopoulo et al., who found that among adult burn patients with acute renal dysfunction, survivors experienced a shorter time between burn and initiation of resuscitation than did nonsurvivors. Holm et al. identified that hypotension and myoglobinuria during resuscitation were more common in patients with early acute renal failure, in comparison with late failure. In a post hoc analysis of data from the Inflammation and Host Response to Injury (Glue Grant) study, patients who received less than 4 mL/kg/TBSA for fluid resuscitation had higher rates of AKI than those who received 4 to 6 mL/kg/TBSA or greater. However, there is a upper limit to the preventative role of fluid resuscitation in AKI: relatively low levels of intraabdominal hypertension (≥12 mmHg) are associated with increased risk of acute renal failure via mechanisms that include renal vein and parenchymal compression.
It is certain that decreased renal perfusion secondary to hypovolemic shock is only one factor in early postburn AKI. Stewart et al. examined the impact of rhabdomyolysis on AKI risk, finding that each 10-fold increase in peak creatine phosphokinase was associated with a 70% increase in the odds of AKI, and an almost 50% increase in the odds of death. Data in burn models are lacking, but in sepsis-associated AKI the following processes were implicated in a recent consensus conference: systemic and renal inflammation, complement activation, renin-angiotensin-aldosterone system dysregulation, mitochondrial dysfunction, and microcirculatory dysfunction.
Gastrointestinal tract
As in the renal circulation, mesenteric vasoconstriction may persist despite apparently adequate resuscitation. One consequence of such ischemia is mucosal barrier dysfunction, which facilitates bacterial and/or endotoxin translocation and predisposes to multiple-system organ dysfunction and to infection. Consistent with this hypothesis, LeVoyer et al. used lactulose/mannose ratios to measure intestinal permeability in patients with large burns. They found that patients who developed early infections had higher ratios than controls or patients who did not develop infections.
In the era before effective antacid therapy, gastroduodenal ulceration (Curling ulcer), beginning within hours of injury, was a common postburn complication, presenting as life-threatening hemorrhage or perforation. , This problem is currently prevented by use of proton-pump inhibitors.
In a 2009 study from the US Army Burn Center that included a large number of combat casualties, ACS and nonocclusive mesenteric ischemia and infarction (NOMI) were the leading causes of abdominal complications. ACS (with or without NOMI) typically presented as a complication of burn resuscitation (see later), whereas NOMI in the absence of ACS typically presented later in the hospital course. The pathophysiology of isolated postburn NOMI is poorly understood. In a prospective observational case-controlled study, NOMI in burn patients was associated with lower cardiac index during the first 24 hours postburn and a higher SOFA score on day 1. The study captured the high mortality (93%) of this complication.
An unanswered question is whether concerns about decreased gastrointestinal perfusion during burn shock should modify the timing of and approach to both enteral resuscitation and enteral nutrition. Arguing from vast experience with enteral resuscitation in children with cholera, it seems reasonable to perform enteral resuscitation (orally or via a tube) using oral rehydration salts, although large-scale randomized controlled trials are lacking. Early enteral nutrition (within the first 24–48 hours of ICU admission) aims to promote the functional integrity of the gut and may be associated with decreased infections and mortality. The safety and efficacy of earlier enteral nutrition in patients with burn shock remains to be determined.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


