Fig. 12.1
Demonstration of the pathogenicity of anti-COL7 autoantibodies in vitro, ex vivo, and in vivo. (a) Schematic diagram of reactive oxygen species (ROS) induced by immune complexes (IC) generated by incubation of anti-COL7 IgG with recombinant COL7. (b) Example of a cryosection of the human skin co-incubated with patient antibodies to COL7 and leukocytes of healthy volunteers. Detachment of the epidermis along the dermal-epidermal junction. (c) Clinical presentation of a C57Bl/6 mouse injected with rabbit anti-COL7 IgG leading to blisters, erosions, alopecia, and scarring. (d) Clinical presentation of an SJL/J mouse immunized with an immunodominant fragment located within the murine NC1 domain showing erosions on the ears and face
12.2.3 Detection of Anti-COL7 Autoantibodies in Other Diseases than EBA
Autoantibodies to COL7 are also detected in other chronic inflammatory diseases; albeit the data on anti-COL7 autoantibody frequencies in these diseases varies greatly. Sometimes, they are detected in patients with inflammatory bowel disease [13–15]. In detail, in patients with ulcerative colitis, frequency may range from 6 to 16 %. In patients with Crohn’s disease, frequency ranges from 4 to 68 %. In addition, anti-COL7 autoantibodies are frequently detected in patients with bullous systemic lupus erythematosus [16]. Frequencies of anti-COL7 autoantibodies in other AIBD are comparatively low [15, 17]: In detail, anti-COL7 antibodies may be detected in patients with bullous pemphigoid (2.6–4 %) and pemphigus vulgaris (up to 9.5 %). In comparison, anti-COL7 autoantibodies are detected in 35 % to almost 100 % of EBA patients and relatively rarely in healthy controls (0–1.6 %) [2, 15, 17, 18]. Furthermore, anti-COL7 autoantibodies are not (except for few case reports) detected in patients with anti-p200 pemphigoid, anti-laminin 332 mucous membrane pemphigoid, celiac disease, or rheumatoid arthritis [13, 17]. The above-described discrepancies in autoantibody frequencies may be due to the use of different diagnostic criteria, detection systems, or a combination thereof.
12.3 Mechanisms Involved in Loss of Tolerance to Type VII Collagen
Up to now, relatively little is known regarding the loss of tolerance by the patient’s immune system to their own collagen VII in EBA. However, both data from EBA patients and animal models point toward a strong genetic influence of EBA susceptibility. In EBA patients, the HLA-DR2 [19] and the HLA-DRB1*15:03 [20] have been reported to be associated with disease susceptibility. Interestingly, the increased prevalence of EBA in Africans [20] and African-Americans [19] points toward a possible contribution of genes outside the MHC locus. These findings have been recently reproduced in immunization-induced EBA: Disease induction in inbred strains of mice showed a significant association of susceptibility with the H2s haplotype [12]. Furthermore, approximately one third of animals from a recently established outbred mouse line [21] are susceptible to immunization-induced EBA. Classical quantitative trait loci (QTL) analysis identified several QTL, located outside the MHC, for EBA susceptibility [22].
In addition to the genetic background, T cells are required for EBA induction. COL7-specific T cells are detected in EBA patients [23] and in mice with immunization-induced EBA [24]. Functional studies underscored the importance of T cells to the loss of tolerance to COL7, as SJLnude mice developed neither anti-COL7 autoantibodies nor clinical disease after immunization with COL7. EBA susceptibility could be restored in these mice by the transfer of T cells from immunized wild-type SJL mice [24]. A detailed comparison of the cytokine milieu in draining lymph nodes of EBA-resistant and EBA-susceptible mice showed a strong Th1 polarization in the latter [25]. This indicates that (1) a Th1 milieu in draining lymph nodes is required for the formation of pathogenic anti-COL7 autoantibodies and (2) possibly certain T-cell subsets are sufficient to mediate the autoantibody production. Inhibition of T-cell function by inhibition of heat shock protein (HSP) 90 impairs skin blistering in immunization-induced EBA and improves disease when used therapeutically in this model [26]. It is, however, currently unknown, which cells present COL7 to the T cells. Mechanisms leading to the loss of tolerance in EBA are summarized in Fig. 12.2.
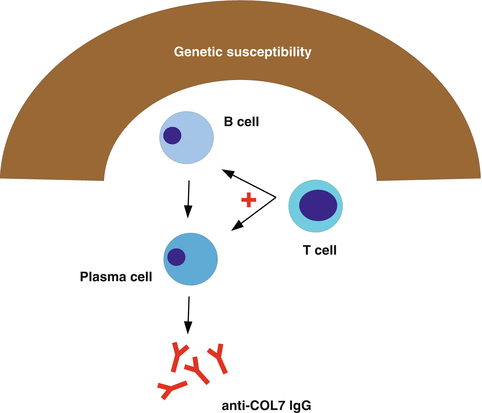
Fig. 12.2
Mechanisms leading to a loss of tolerance to COL7. Individuals with an EBA-susceptible genetic background (including genes inside and outside the MHC locus) generate anti-COL7 autoantibody-producing plasma cells. Establishing the autoantibody response requires (predominantly Th1 polarized) T cells
12.4 Autoantibody-Induced Tissue Injury in EBA
12.4.1 Biodistribution and Half-Life of Anti-COL7 Autoantibodies
In two large EBA patient cohorts, IgG anti-COL7 antibodies were detected in 72 %. The anti-COL7 IgG antibodies were of all IgG subclasses, with the following detection frequency: IgG4>IgG1>IgG2>IgG3. In addition to IgG, IgA autoantibodies solely or in combination with IgG antibodies are detected in 53 % of the patients [2, 18, 27]. In humans, COL7 expression has been described to be restricted to the basement membrane zone beneath stratified squamous epithelia. However, while it is predominantly expressed in the skin, COL7 expression can also be detected along the entire gastrointestinal tract, with a high expression in the oral cavity and esophagus, but a low expression in the colon. Binding of anti-COL7 autoantibodies parallels the antigen’s expression [13, 28–31]. In mice, the half-life of anti-COL7 IgG autoantibodies was shown to be controlled by the neonatal Fc receptor (FcRn), as FcRn deficiency was partially protected from antibody transfer-induced EBA [32]. Continuous observation of serum and tissue-bound anti-COL7 IgG in mice after transplacental transfer of the IgG showed persistence of anti-COL7 IgG in the circulation for 4 weeks. In contrast, the presence of tissue-bound autoantibodies was observed for a total of 8 weeks [33]. In a case of neonatal EBA from a mother with EBA in remission, the blistering in the neonate lasted for 7–8 weeks [34].
12.4.2 Epitopes Targeted by Anti-COL7 Autoantibodies
Epitope mapping studies showed that the majority of autoantibodies from EBA patients target epitopes located within the noncollagenous (NC)-1 domain of COL7 [35–38]. Rarely epitopes within the NC2 [39] or the collagenous domains [40, 41] are recognized. Subsequent epitope mapping confirmed these findings, all pointing to a mainly NC1-mediated autoantibody response in EBA patients [42]. With a few exceptions [11, 43], the pathogenic relevance of these autoantibodies has not yet been demonstrated. At least for the inflammatory variant of EBA, it is tempting to speculate that autoantibody deposition at the dermal-epidermal junction will lead to blister formation independent of the targeted COL7 epitope. Regarding the noninflammatory EBA manifestations, the target epitope may be of relevance, as autoantibody binding may interfere with the assembly of intermolecular structures within the basement membrane.
12.4.3 Generation of a Proinflammatory Milieu in the Skin
After binding of anti-COL7 autoantibodies to the dermal-epidermal junction of the skin, a proinflammatory milieu is created. So far, complement deposition along the dermal-epidermal junction has been identified as the first molecular event that takes place after IgG deposition at the dermal-epidermal junction [9, 28]. In line with this observation, mice lacking C5 expression were completely protected from EBA induction by anti-COL7 IgG transfer [9]. Further investigations showed that complement is predominantly but not exclusively activated by the alternative pathway [44]. Interestingly, the contribution of the complement system may be more complex than suggested by these initial studies, as we recently observed the induction of experimental EBA in the absence of complement deposition; vice versa, mice with complement deposits at the dermal-epidermal junction did not develop clinical disease [22]. These findings are supported by recent observations in a humanized mouse model of bullous pemphigoid, where tissue injury can also be induced by both complement-dependent and complement-independent pathways [45]. Therefore, other pathways may be operative initiating the extravasation of effector cells into the skin. Possibly, this process is mediated by resident immune cells, i.e., mast cells and/or macrophages, as described in animal models of bullous pemphigoid [46]. At the molecular level, cytokines are likely involved, as an aberrant cytokine expression has been described in several AIBD [47] (Fig. 12.3).
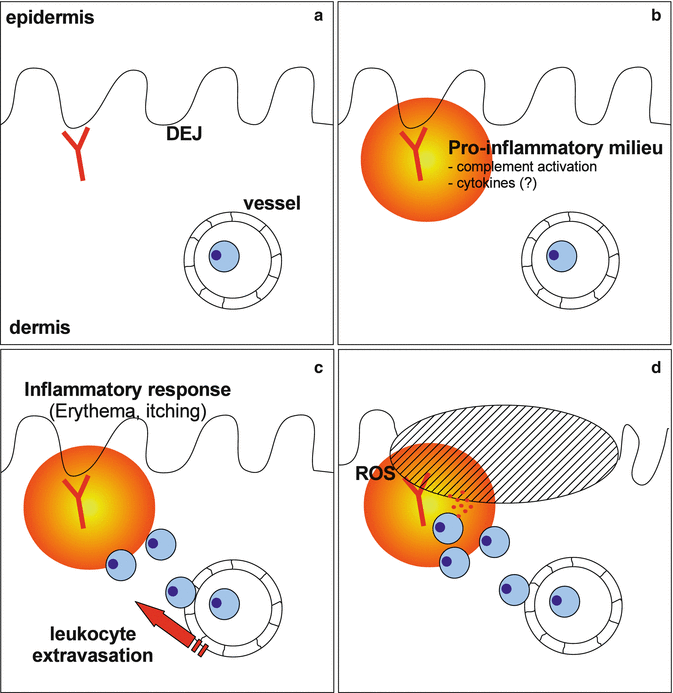
Fig. 12.3
Autoantibody-induced tissue injury in EBA. (a) Tissue injury is initiated by autoantibody binding to the dermal-epidermal junction (DEJ). In experimental EBA, autoantibody deposition can be detected within the first 24–48 h after autoantibody transfer by direct immunofluorescence microscopy. (b) Autoantibody deposition leads to the generation of a proinflammatory milieu in the skin (yellow circle) and an inflammatory response. (c) Subsequently, a CD18-dependent leukocyte migration into the skin is seen. (d) Ultimately, Fc gamma receptor-dependent mechanisms lead to the activation of extravasated effector leukocytes, e.g., neutrophils. This activation results in the release of reactive oxygen species (ROS) and proteolytic enzymes (red dots), which then cause dermal-epidermal separation
12.4.4 Extravasation of Effector Cells into the Skin
This proinflammatory milieu leads to the extravasation of effector cells into the skin. Presumably, albeit not fully experimentally shown, this process is mediated by an increased expression of endothelial adhesion molecules and an activation of leukocyte integrins. Direct experimental evidence demonstrates that leukocyte beta2 integrins are indispensable for blister formation, as CD18-deficient mice were completely protected from blister induction by anti-COL7 IgG transfer [48]. The importance of beta2 integrins for autoantibody-induced tissue injury has also been demonstrated in a neonatal mouse model of bullous pemphigoid, where blockade of CD18 led to a complete protection of mice from disease induction [49]. For all other steps of leukocyte extravasation, i.e., rolling and transmigration [50], no direct experimental data is available in EBA, whereas an increased expression of other adhesion molecules and cytokines has been noted in patients with pemphigoid [47, 51, 52] (Fig. 12.3).
12.4.5 Fc-Dependent Activation of Effector Cells
Once migrated into the skin, effector leukocytes bind to the immune complexes located at the dermal-epidermal junction. This process is both Fc and Fc gamma receptor dependent. Anti-COL7 F(ab) fragments bind to COL7 both in vitro and in vivo, but fail to induce blister formation in cryosections of human skin [6] and in mice [9, 53]. In line, avian IgY, which does not bind to murine Fc gamma receptors, does not induce blisters when injected into mice [54]. Collectively, these data point to a significant contribution of activating Fc gamma receptors. We recently provided direct experimental evidence for this assumption. In detail, mice lacking expression of the common gamma chain of murine activating Fc gamma receptors were completely protected from EBA induction by transfer of anti-COL7 IgG. By use of knockout mice and function-blocking antibodies, we showed that this autoantibody-induced tissue damage in EBA solely depends on Fc gamma RIV expression. All other known activating murine Fc gamma receptors, including Fc gamma RI and RIII, were not required to induce blisters [55]. These proinflammatory effects of activating Fc gamma receptors were counteracted by mechanisms mediated by inhibitory Fc gamma RIIB. Mice deficient for Fc gamma RIIB showed a significantly enhanced EBA phenotype after receiving anti-COL7 IgG [55] (Fig. 12.3). Interestingly, a different Fc gamma receptor usage has been described in bullous pemphigoid (BP) in neonatal mice. In this model, Fc gamma RIII played a critical role in the activation of infiltrating neutrophils and subsequent skin blistering. Furthermore, lack of Fc gamma RIIB expression had no impact on disease manifestation [56]. Future work will have to define if this difference is due to the different ages of mice (adult vs. neonatal) or the underlying disease.
12.4.6 Mediators of Tissue Injury
Neutrophils have been identified as the main effector cells in experimental EBA, as neutrophil depletion completely protects mice from autoantibody transfer-induced disease [48]. The antibody used for neutrophil depletion in this study, however, also leads to depletion of other cell types, i.e., monocytes and macrophages. Hence, a contribution of these cells cannot be excluded at this point.
After binding of effector cells to immune complexes located at the dermal-epidermal junction, signaling events downstream of ITIM lead to neutrophil activation. So far, several signaling molecules have been identified to contribute to neutrophil activation induced by immune complexes of autoantibodies and hemidesmosomal proteins. Among these, phosphoinositide 3-kinase beta (PI3Kβ[beta]) has been characterized in most detail: In vitro, at low doses of immune complexes, loss of PI3Kβ[beta] alone substantially inhibited reactive oxygen species production. At higher doses of immune complexes, similar suppression was achieved only by targeting both PI3Kβ[beta] and PI3Kδ[delta], suggesting this pathway displays a stimulus strength-dependent redundancy. In vivo, induction of experimental EBA by autoantibody transfer was significantly impaired in PI3Kβ[beta]-deficient mice. By use of bone marrow chimeric mice, it was shown that this protection is autonomous to the hematopoietic lineage, as irradiated wild-type C57Bl/6J mice reconstituted with bone marrow from PI3Kβ[beta]-deficient mice were similarly protected from the development of blister formation [8]. More recently, we also identified a crucial contribution of p38, Akt, and ERK1/2 phosphorylation in immune-complex activation of neutrophils. Inhibition of p38, Akt, and ERK1/2 phosphorylation blocked release of reactive oxygen species and/or degranulation of immune complex-activated neutrophils and impaired autoantibody- and neutrophil-dependent dermal–epidermal separation. In line with these in vitro findings, blockade of either p38 or ERK1/2 phosphorylation impaired induction of experimental EBA by autoantibody transfer [57].
These signaling events ultimately lead to neutrophil activation, which, among other biological effects, is characterized by release of reactive oxygen species and proteolytic enzymes. Pharmacological inhibition or deficiency of human NADPH oxidase abolishes dermal-epidermal separation caused by anti-COL7 autoantibodies and granulocytes ex vivo. In line, neutrophil cytosolic factor 1-deficient mice are protected from experimental EBA, and transfer of neutrophil cytosolic factor 1-sufficient granulocytes into neutrophil cytosolic factor 1-deficient mice reconstitutes disease susceptibility [48]. Furthermore, expression profiling revealed an increased expression of matrix metalloproteinase (MMP)-8 (neutrophil collagenase) and MMP-13 (collagenase-3) in the skin of mice with experimental EBA [55]. Functional studies in cryosections of human skin showed that inhibition of elastase and gelatinase B completely abolished the blister-inducing capacity of neutrophils ex vivo [58].
Interestingly, the actin-remodeling protein flightless I (Flii) has recently been demonstrated to have an important role in mediating cellular adhesion in EBA. Previous work in wound healing models had shown that reducing Flii expression improved wound healing, while Flii overexpression resulted in delayed wound closure, increased skin fragility, and impaired wound healing [59]. Similar findings were observed in experimental EBA, where induction of disease led to an increased cutaneous Flii expression. In line, reduced Flii expression in Flii+/− mice significantly impaired the induction of experimental EBA [60]. The events leading to tissue injury in EBA are summarized in Fig. 12.3.
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Azathioprine
Azathioprine
 COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


