Major criteria
Polymorphous mucocutaneous eruption
Painful and persistent stomatitis
Respiratory involvement
Concurrent internal neoplasia
Immunoprecipitation: 190, 210 (doublet), 230, and 250 kDa bands
Minor criteria
Acantholysis and/or subepidermal split
Direct immunofluorescence showing both intercellular and linear basement membrane staining of the epidermis
Indirect immunofluorescence staining of rodent bladder epithelium
Lack of correlation of mucocutaneous disease with anti-Dsg 1 and anti-Dsg 3 antibodies
There have been over 400 cases of PAMS reported in the literature. There is a slight male predominance with most cases occurring between 45 and 70 years of age. Interestingly, there is a significant association between patients with paraneoplastic autoimmune multiorgan syndrome and HLA-class II DRB1*03 and HLA-Cw*14 [5, 6]. This differs from HLA associations in pemphigus vulgaris and foliaceous which are strongly associated with HLA-class II DRB1+4 and DRB1*14.
Most cases of PAMS occur in association with a lymphoproliferative disorder. In a review of 163 cases, non-Hodgkin’s lymphoma (38.6 %), chronic lymphocytic leukemia (18.4 %), Castleman’s disease (18.4 %), and thymoma (5.5 %) are the most commonly reported concurrent neoplasms [7]. Castleman’s disease appears to be reported in much higher frequency in children [2, 8, 9] and in Chinese PAMS (77 %) [10]. Of several cases reported to occur in association with thymoma and Castleman’s disease, further pathologic examination revealed concurrent follicular dendritic cell sarcoma (FDCS) [11, 12]. Follicular dendritic cells are non-phagocytic and non-lymphoid accessory cells of the immune system which are essential for antigen presentation and regulation of the immune responses in the lymph node germinal centers [13, 14]. Terminal deoxynucleotidyl transferase (TdT)-positive T cells have been identified in FDCS that may serve to amplify CD8+ cytotoxic T-lymphocyte (CTL) responses and potentially autoimmunity, including autoreactive T cells directed against lung and other epithelial antigens [15–17]. This provides a potential novel understanding applicable to mechanisms underlying autoimmunity. Additionally, Wang et al. have shown autoreactive B-cell clones produce autoantibodies against plakin proteins in FDCS, Castleman’s disease, as well as thymomas [12, 18]. These results suggest that the presence of B-lymphocyte clones may be a common mechanism in PAMS associated with various tumors.
25.2 Clinical Features
PAMS presents with a polymorphous cutaneous eruption that has been reported on a spectrum ranging from blisters, erosions, patches, papules, and plaques. Several subsets have been described: (1) bullous pemphigoid-like [19–21], (2) pemphigus-like [1, 22], (3) pemphigus vegetans-like [23, 24], (4) erythema multiforme-like [9, 11, 25], (5) graft-versus-host disease-like [26], and (6) lichen planus-like [25, 27, 28]. Severe, more recalcitrant oral mucosal lesions are the hallmark of the disease and usually are the initial manifestation [29].
Pemphigus vulgaris fundamentally differs clinically from PAMS in several ways (Fig. 25.1). PAMS may affect any cutaneous surface including the palms and the soles, and lesions tend to be polymorphous and not limited to flaccid bullae and erosions as seen in pemphigus vulgaris. While both have oral mucosal disease, in pemphigus vulgaris oral mucosal lesions are discrete and more demarcated with areas of sparing. In PAMS, the oral involvement is more diffuse, resulting in a generalized erosive stomatitis. In addition to conjunctival and esophageal mucosa, PAMS may also affect gastric, duodenal, respiratory, colonic, and urogenital epithelia that are spared in pemphigus vulgaris [30, 31]. Affected respiratory mucous membranes in PAMS lead to bronchiolitis obliterans, resulting in rapidly progressive and often fatal respiratory failure characterized by deposition of Ig and complement in pulmonary tissue [4, 32]. Furthermore, PAMS has been reported to occur in association with glomerulonephritis and a paraneoplastic neurologic syndrome, further expanding the multiorgan involvement of this disease [33]. PAMS has an overall mortality of 90 % despite therapy, as opposed to that of pemphigus vulgaris, which is now about 5–7 % with therapy. Most cases of PAMS occur in association with a lymphoproliferative disorder, but at the time of presentation, approximately a third of patients have no known or identified malignancy [19].
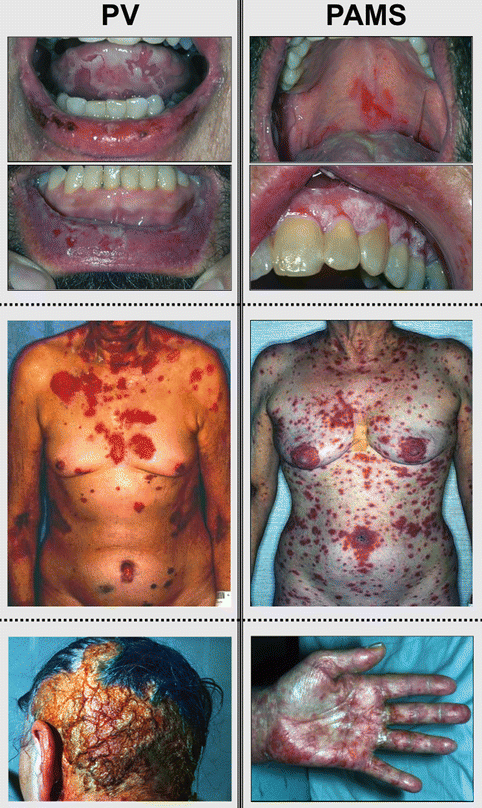
Fig. 25.1
Typical mucocutaneous manifestations of pemphigus vulgaris versus paraneoplastic autoimmune multiorgan syndrome (Modified from Czernik et al. [29])
PAMS is not infrequently misdiagnosed as chronic, erosive stomatitis or atypical erythema multiforme and, in more severe cases, as Stevens-Johnson syndrome. Severe, more generalized cases may be referred to burn centers or intensive care units before the correct diagnosis is established [34]. It is imperative to perform a complete and thorough medical examination to uncover potential underlying neoplastic or malignant conditions. Skin biopsy for routine histology and immunopathology as well as serologic screening should be included in the laboratory evaluation (see below) of suspected patients with PAMS to avoid overlooking the correct diagnosis, pursuing indicated imaging studies, and instituting appropriate therapies.
Respiratory failure is one of the most common causes of death in PAMS. Patients develop a constrictive and obstructive pattern of respiratory failure with features of bronchiolitis obliterans and a functional pattern of obstructive or restrictive bronchiolitis similar to that observed in allogenic lung transplantation [35]. In our recent review of pulmonary involvement at the Mayo Clinic [36], two thirds of patients with constrictive bronchiolitis expired during the follow-up period, in contrast to only one third of those without this type of pulmonary disease. Resection of the neoplasm or induction of a complete response in cases of malignancy, despite resolution of other mucocutaneous features of PAMS, often does not change the rapid, inexorable decline and fatal course of the progressive respiratory failure.
25.3 Histopathology
The histopathologic hallmark is an interface reaction pattern, characterized by basal cell vacuolar degeneration and dyskeratotic and necrotic keratinocytes with lymphocytic inflammation and lymphocytic exocytosis. The reaction pattern may include either scattered lymphocytes at the BMZ (interface vacuolar reaction pattern) or a band-like infiltrate in the upper dermis (interface lichenoid reaction pattern). Intraepidermal blisters and subepidermal blisters with acantholysis may be present and are less prominent features than in pemphigus vulgaris or bullous pemphigoid [37]. Postmortem pulmonary specimens in PAMS demonstrate individual cells and clusters of epithelial cells in the tracheal, bronchial, and small-airway lumen with complete plugging of the alveoli [4].
25.4 Pathophysiology
Both humoral and cytotoxic immunities are involved in the development of PAMS (Fig. 25.2). Passive transfer of immunoglobulins isolated from patients with PAMS produced mucocutaneous supra-basilar acantholysis in mice providing evidence that these antibodies are pathogenic [1]. Exposure of epithelial cells to autoantibodies against the linker subdomain of plakin proteins envoplakin and periplakin from PAMS sera leads to internalizations of these antibodies and retraction of keratin filaments [38]. Both these observations point to the pathogenesis of autoantibodies in the development of PAMS. In fact, cell cultures from resected Castleman’s disease, thymoma, and follicular dendritic cell sarcoma have all been shown to produce autoantibodies to desmosomal and hemidesmosomal proteins [18, 39].
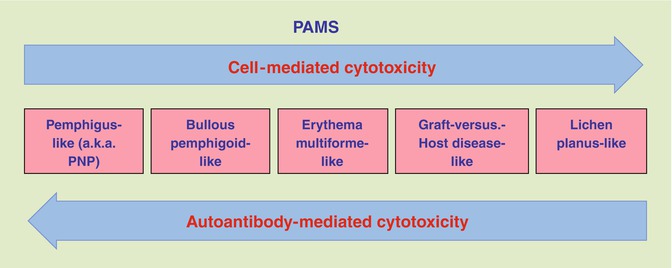
Fig. 25.2
Interrelationships between the predominant immunopathological mechanism of target cell damage and the clinicopathological form of PAMS
Deposition of polyclonal IgG autoantibodies and complement is found in multiple organ systems including mucocutaneous, conjunctival, bronchial, muscle, urinary bladder, and renal glomeruli [4]. Immunoglobulins produce three distinct or overlapping staining patterns: (1) fishnet-like intercellular staining of the epithelium (pemphigus-like), (2) linear staining at the basement membrane zone (pemphigoid-like), or (3) homogenous within the cells (apoptosis-like). Serum antibodies have been detected by indirect immunofluorescence technique utilizing a variety of epithelial and non-epithelial tissue substrates including monkey esophagus (86 % sensitivity), murine tongue (100 % sensitivity), and mouse bladder (75 % sensitivity) [40].
Immunoprecipitation has detected circulating autoantibodies that recognize keratinocyte proteins of various molecular weights: 40, 60, 70, 80, 95, 105, 130, 150, 170, 190, 210 (doublet), 230, and 250 kDa [1, 4]. Most sera recognize members of the plakin family including desmoplakin I, desmoplakin II, BPAG1, envoplakin, periplakin, and plectin. Antibodies against envoplakin and periplakin are consistently recognized and believed to be the most sensitive and specific indicators of disease [41]. In some cases, decline of envoplakin and desmoglein 3 antibodies has been associated with improved clinical response in benign tumors, such as thymoma, treated with prednisone [42]. However, the development of anti-keratinocyte autoantibodies has been reported as delayed in onset, limited, or absent in a number of cases [27, 43, 44]. The 170 kD antigen target has been identified as a protease inhibitor, alpha-2 macroglobulin-like (A2ML1) protein, though its pathogenic role in disease development and tissue injury remains unknown [45].
The role of desmoglein antibodies in the pathogenesis of PAMS is controversial. An increasing number of reports indicate that these antibodies are present in only a minority of cases and, when present, are not consistently pathogenic [4, 46–49]. Indirect immunofluorescence studies on COS-7 cells transiently transfected with desmocollin 1–3 cDNA demonstrated the presence of serum IgG and IgA antibodies to desmocollin 3, as well as IgG antibodies to desmocollin 2 [28]. Therefore, humoral immunity in PAMS, unlike other autoimmune blistering diseases, is more heterogeneous, inconsistently positive, and not completely understood.
Cytotoxic cell-mediated immunity and antibody-dependent cellular cytotoxicity (ADCC) are involved in the pathogenesis of PAMS. Cytotoxic T lymphocytes (CTL), NK cells, and monocytes and macrophages localize to the epidermal-dermal junction [4]. Immunohistochemical staining shows mostly CD8+ cytotoxic T lymphocytes within the basal and suprabasal epidermis, juxtaposed to apoptotic keratinocytes. Additionally, CD68+ monocytes/macrophages (effectors of natural cytotoxicity) surround the subepidermal clefts. The infiltrate is similar to other inflammatory, autodestructive dermatoses characterized by keratinocyte apoptosis, erythema multiforme, GVHD, and lichen planus.
Interferon gamma and tumor necrosis factor appear to be central to the development of the graft-versus-host disease-like form of PAMS [50]. Infiltrating CD68+ monocytes and macrophages stain for inducible nitric oxide synthase (iNOS) in PAMS indicating they are activated and involved in the pathogenesis of PAMS [4]. Nitric oxide may increase Fas-induced apoptosis in basal keratinocytes, explaining one possible mechanism for the loss of adhesion within the tegumental epithelium [51].
Epitope spreading, the development of autoimmunity following exposure to previously hidden self-antigens, is also suggested by several observations. At the time of initial presentation, immunofluorescence studies may be negative, reflecting a lack of humoral immunity. In one recently published case, indirect immunofluorescence identified autoantibodies which reacted with rat bladder epithelium consistent with PAMS [52]. These antibodies, however, were not detectable until 1 year following the initial clinical presentation.
The heterogeneous clinicopathological presentation is likely a direct reflection of the diverse immunologic mechanisms of autoimmunity involving diverse effecters of humoral and cellular cytotoxicity. The interrelationships between the predominant immunopathological mechanism of target cell damage and clinical presentation of PAMS are shown in Fig. 25.2. Bullous eruptions of PAMS have a predominant humoral response as originally reported by Anhalt [1], where passive transfer of PAMS sera led to suprabasal acantholysis in mice. Lichenoid eruptions have been exclusively linked to cytotoxic infiltrates in the absence of detectable antibodies [27]. The spectrum of PAMS likely includes patients with disease predominantly or exclusively mediated by CTL and others predominantly by autoantibodies.
25.5 Management
Clinical, histopathologic, and immunologic criteria should ideally be met in order to firmly establish the diagnosis. In patients without a known malignancy, a complete blood count; serum protein electrophoresis; computerized tomography of the chest, abdomen, and pelvis; and diagnostic biopsies (i.e., bone marrow, lymph nodes, or solid tumor), as indicated, should be obtained.
Treatment is difficult and the best outcomes have been reported in cases of resectable tumors [4, 53, 54]. Complete resection of 20 cases of neoplasia associated with PAMS led to sustained mucocutaneous improvement in 15 patients; however, respiratory symptoms and compromised pulmonary function persisted in the majority of these cases [53]. Pulmonary involvement is often progressive and irreversible despite tumor resection and treatment of mucocutaneous disease. However, it appears that some cases of constrictive bronchiolitis exhibit a more protracted and less aggressive course. In the cases when the neoplasm was resected early and immunosuppressive treatment, including parenteral corticosteroids, cyclosporine, cyclophosphamide, and methotrexate, was timely initiated, the respiratory disease was substantially slowed and residual pulmonary function stabilized. Nonetheless, destruction of the lung airways and development of fibrosis and obstruction are usually nonreversible and result in chronic supplemental oxygen requirement. In these cases, lung transplantation may be the only viable option for intervention.
In unresectable disease, treatment of the underlying malignancy is often not associated with disease improvement [54, 55]. These patients are often resistant to all conventional therapies. A combination of prednisone (0.5–1 mg/kg) and cyclosporine (5 mg/kg) ± cyclophosphamide (2 mg/kg) may be the most appropriate pharmacotherapy [56]. In addition there have been numerous case reports showing promise with additional agents including immunoablative high-dose cyclophosphamide without stem cell rescue, immunoapheresis, intravenous immunoglobulin, rituximab, alemtuzumab, as well as pyridostigmine bromide [54, 55, 57–62]. Overall, prognosis is poor and PAMS may have a fatality rate as high as 90 % [32]. The majority of patients succumb to sepsis, respiratory failure, or the underlying neoplasm [55].
In conclusion, PAMS is a multiorgan autoimmune syndrome affecting both tegumental epithelium and internal organs. Patients with PAMS differ distinctively, though at times more subtly from patients with classical pemphigus, and may have lesions that resemble not only pemphigus but also pemphigoid, erythema multiforme, lichen planus, lichen planus pemphigoides, and graft-versus-host disease. Mucocutaneous lesions in patients with PAMS occur as a result of both humoral and cell-mediated immune mechanisms. Cell-mediated cytotoxicity in PAMS involves both autoreactive lymphocytes and circulating autoreactive IgGs that recruit peripheral blood mononuclear cells into antibody-dependent cellular cytotoxicity against self-epithelium. Sloughing of bronchial epithelial cells can contribute to occlusion of the small airways that provides a likely pathologic mechanism for the respiratory failure that constitutes a terminal event in many patients with PAMS [4]. Treatment is difficult, and PAMS often does not respond to therapies directed toward the underlying malignancy. Based on these more recent and clinically better defined understandings of the disease, new therapeutic approaches with more potent pharmaceutical agents targeting the immune system should be developed to improve the often fatal course in patients with PAMS.
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


