Abstract
The parapsoriasis group of skin disorders includes small and large plaque parapsoriasis, with some clinicians extending the spectrum of T-cell related disorders to include acute and chronic forms of pityriasis lichenoides and even lymphomatoid papulosis. These are all chronic, idiopathic dermatoses characterized by T-cell infiltrates that often exhibit dominant clonality. They have a variable association with overt lymphomas. Pityriasis rubra pilaris is an idiopathic disorder with six types, including various juvenile and adult forms as well as an HIV-associated type (VI). Pityriasis rosea is a self-limited eruption more commonly seen in adolescents and young adults with oval papulosquamous lesions following Langer lines. Pityriasis rotunda is a rare condition in which large circular lesions without obvious inflammation are present, often in association with malnutrition. Granular parakeratosis is an uncommon condition where coalescing keratotic papules develop within major body folds, most frequently the axillae of adult women.
Keywords
parapsoriasis, digitate dermatosis, pityriasis lichenoides, Mucha–Habermann disease, pityriasis rubra pilaris, pityriasis rosea, pityriasis rotunda, granular parakeratosis, small plaque parapsoriasis, large plaque parapsoriasis, pityriasis lichenoides et varioliformis acuta, pityriasis lichenoides chronica
Small Plaque Parapsoriasis
▪ Parapsoriasis en plaques ▪ Chronic superficial [scaly] dermatitis ▪ Digitate dermatosis (variant) ▪ Xanthoerythrodermia perstans (variant)
- ▪
Chronic, asymptomatic, erythematous scaly patches
- ▪
Lesions are generally <5 cm in diameter or digitate
- ▪
Histologically, mild, nonspecific spongiotic dermatitis with parakeratosis is seen
- ▪
There is a predominance of CD4 + T cells in the lymphocytic infiltrate
- ▪
A dominant T-cell clonality is demonstrable in some cases
Introduction
Small plaque parapsoriasis is an idiopathic, chronic dermatosis classified within the parapsoriasis group of skin diseases, which also includes large plaque parapsoriasis. Some authors include the acute and chronic variants of pityriasis lichenoides and even lymphomatoid papulosis within the spectrum of clonal T-cell related dermatitides. This group is significant for the tendency of its disorders to coexist or overlap with one another and for its association with lymphomas . Of note, there is a school of thought that large plaque parapsoriasis represents the earliest stage of mycosis fungoides .
History
The parapsoriasis group was defined by Brocq in 1902 and refined subsequently by several authors whose work was summarized by Lambert and Everett in 1981 . Although he recognized that these disorders were distinct from psoriasis, Brocq chose the term parapsoriasis to reflect overlapping clinical features including chronicity, idiopathic origin, recalcitrance to contemporary therapies, and lesions that were often nonpruritic. It then became widely accepted that small plaque parapsoriasis and large plaque parapsoriasis were distinct entities , and that large plaque parapsoriasis and its variants were closely related to the patch stage of mycosis fungoides .
Epidemiology
Small plaque parapsoriasis, like large plaque parapsoriasis, is more common in middle-aged and elderly individuals, but may also occur in children. Parapsoriasis peaks during the fifth decade of life and occurs in all racial groups and geographic regions. There is a male predominance that is greater for small plaque parapsoriasis (male : female ratio ~3 : 1) than it is for large plaque parapsoriasis.
Pathogenesis
The etiology of small plaque parapsoriasis is unknown. It is characterized by superficial cutaneous lymphoid infiltrates composed primarily of CD4 + T cells . Dominant T-cell clonality has been demonstrated in some cases of small plaque parapsoriasis . This variability may be an artifact of a relatively low T-cell density. Alternatively, it may be that some cases are truly polyclonal and represent a distinct biologic subset of disease. The term clonal dermatitis has been coined to reflect an intermediate or transitional step between chronic dermatitis and overt CTCL. Small retrospective studies suggest that patients with “clonal dermatitis”, which also includes large plaque parapsoriasis, have an ~20% risk of progression to overt CTCL over 5 years . However, it is important to note that some cases of CTCL may contain oligoclonal T cells from which an overt dominant T-cell clone has yet to evolve .
Clinical Features
Despite inclusion of the word “plaque” in their terminology or use of the term parapsoriasis en plaques , both small plaque and large plaque parapsoriasis are diseases composed of patches rather than plaques. In both entities, skin lesions are chronic and generally asymptomatic or mildly pruritic. They may wax and wane early on during their clinical course, but they typically become persistent and can slowly progress to become more extensive. Some cases regress fully, either spontaneously or after therapy, but this may require a period of several years. Lesions can be widespread on the trunk and extremities or more limited in their distribution. In the latter instance, they often favor more sun-protected sites.
Small plaque parapsoriasis typically presents as round–oval patches that are generally <5 cm in diameter ( Fig. 9.1A ). They are variably erythematous (but often less intense than psoriasis) and covered with a fine scale. Those with a yellow hue have been termed “xanthoerythrodermia perstans” . An important variant of small plaque parapsoriasis is “digitate dermatosis”, which presents as elongated, finger-like patches symmetrically distributed on the flanks ( Fig. 9.1B ). These lesions are an exception to the 5-cm rule in that they may measure 10 cm or more along their long axis. The reported risk of progression to mycosis fungoides varies from zero to low .
Pathology
Patches of small plaque parapsoriasis exhibit a mild, nonspecific spongiotic dermatitis with focal parakeratosis. Exocytosis of lymphocytes is variable but usually present.
Differential Diagnosis
The diagnosis of either small plaque parapsoriasis or large plaque parapsoriasis is based on a correlation between clinical and histopathologic findings; other tests are of minimal value. They are distinguished from each other by their clinical features. The principal entities in the differential diagnosis are listed in Table 9.1 . Both small plaque and large plaque parapsoriasis are differentiated from mycosis fungoides on the basis of failing to meet the minimum histopathologic criteria for the latter disorder.
| PRINCIPAL DIFFERENTIAL DIAGNOSIS OF SMALL PLAQUE PARAPSORIASIS AND LARGE PLAQUE PARAPSORIASIS | |
|---|---|
| Small plaque parapsoriasis | Large plaque parapsoriasis |
|
|
Pityriasis rosea is distinguished from small plaque parapsoriasis by the presence of a herald patch and spontaneous resolution within a few months. Psoriasis and secondary syphilis are excluded on the basis of clinicopathologic and serologic features. Patients with pityriasis lichenoides, acute and chronic, generally have lesions that are smaller and more widely distributed than do patients with small plaque parapsoriasis. Drug eruptions resembling pityriasis rosea or mycosis fungoides must also be excluded by clinicopathologic correlation.
Treatment
All therapeutic approaches are based primarily on uncontrolled case series, case reports or anecdotal evidence ( Table 9.2 ). In this author’s opinion (GSW), patients with small plaque parapsoriasis should be reassured that their risk of developing mycosis fungoides is practically a nonissue. Ackerman and co-workers disagree and have expressed the opinion that even small plaque parapsoriasis is essentially a forme fruste of mycosis fungoides. Patients with small plaque parapsoriasis may be followed without treatment if they prefer. Standard therapy includes topical corticosteroids, topical coal tar products, and various forms of phototherapy. The role of newer topical agents, such as bexarotene, calcineurin inhibitors and imiquimod, remains to be established. There is some concern that calcineurin inhibitors might be implicated if lymphoma were to develop. Until this issue is clarified, due caution should be exercised in the treatment of small plaque parapsoriasis.
Large Plaque Parapsoriasis
▪ Parapsoriasis en plaques ▪ Retiform parapsoriasis (variant) ▪ Parakeratosis variegata (variant)
- ▪
Chronic, asymptomatic, erythematous scaly patches
- ▪
Lesions are generally >5 cm in diameter
- ▪
Histologically, a nonspecific spongiotic dermatitis or an interface lymphocytic infiltrate with a variable degree of lichenoid features is seen
- ▪
There is a predominance of CD4 + T cells in the lymphocytic infiltrate
- ▪
A dominant T-cell clonality is demonstrable in many cases
Introduction, History, and Epidemiology
Large plaque parapsoriasis is an idiopathic, chronic dermatosis classified within the parapsoriasis group of skin diseases. Its history and epidemiology are discussed in the previous section.
Pathogenesis
The etiology of large plaque parapsoriasis is unknown. It is characterized by superficial cutaneous lymphoid infiltrates composed primarily of CD4 + T cells . Dominant T-cell clonality has been demonstrated in many patients with large plaque parapsoriasis . It is now well established that at least some cases of large plaque parapsoriasis represent the patch stage of mycosis fungoides (see Ch. 120 ). This helps to explain the progression of large plaque parapsoriasis to more overt forms of lymphoma at a rate of 10% to 35% over a period of 6–10 years .
Clinical Features
Large plaque parapsoriasis presents as variably erythematous, round or irregularly shaped, scaly patches that are >5 cm in diameter ( Fig. 9.2A ). They may or may not exhibit the clinical triad of atrophy, telangiectasia, and hyper/hypopigmentation that warrants the descriptive designation “poikiloderma vasculare atrophicans”. Rather than being a specific entity, poikiloderma can be seen in several disorders, including certain autoimmune connective tissue diseases (e.g. dermatomyositis) and genodermatoses, in addition to large plaque parapsoriasis and mycosis fungoides. “Retiform parapsoriasis”, also known as “parapsoriasis variegata”, “parakeratosis variegata” or “parapsoriasis lichenoides”, is a variant of large plaque parapsoriasis in which there are widespread, ill-defined patches in a net-like or zebra-stripe pattern ( Fig. 9.2B ). Long-term follow-up has shown that virtually all of the patients with these rare retiform variants progress to overt mycosis fungoides.
Pathology
Lesions of large plaque parapsoriasis may exhibit parakeratosis and a mild, nonspecific spongiotic dermatitis or an interface lymphocytic infiltrate with a variable degree of lichenoid features. A number of cases of large plaque parapsoriasis are indistinguishable from the patch stage of mycosis fungoides and may contain atypical lymphoid cells (see Ch. 120 ). While these cases should be diagnosed as mycosis fungoides, their proportion is difficult to establish because experts disagree on the minimum criteria required to histopathologically diagnose mycosis fungoides. The term large plaque parapsoriasis should be used for those remaining patients who fail to show the histopathologic features of mycosis fungoides. However, repeat biopsies are recommended, as the histopathologic features may be rendered nonspecific by previous treatments and findings may vary amongst different lesions sampled at the same time. Poikilodermatous variants of large plaque parapsoriasis demonstrate the histopathologic triad of epidermal atrophy, telangiectasia, and pigment incontinence.
Differential Diagnosis
Large plaque parapsoriasis and small plaque parapsoriasis are distinguished from each other by their clinical features. The major entities in the differential diagnosis are listed in Table 9.1 . Some patients with lesions clinically consistent with large plaque parapsoriasis will meet microscopic criteria for mycosis fungoides, but others will not. Therefore, large plaque parapsoriasis remains a useful designation for the latter group.
Treatment
All therapeutic approaches are based primarily on uncontrolled case series, case reports or anecdotal evidence (see Table 9.2 ). Patients with large plaque parapsoriasis should be treated regardless of whether one believes that their lesions already represent or may evolve into mycosis fungoides. Initial therapy may be similar to that described for small plaque parapsoriasis. Those patients with large plaque parapsoriasis in whom the histopathologic criteria for mycosis fungoides are met can be offered the therapeutic options outlined in Table 120.5 .
Pityriasis Lichenoides et Varioliformis Acuta and Pityriasis Lichenoides Chronica
▪ Mucha–Habermann disease (PLEVA) ▪ Guttate parapsoriasis (PLC)
- ▪
Pityriasis lichenoides et varioliformis acuta (PLEVA) and pityriasis lichenoides chronica (PLC) are two ends of a disease spectrum
- ▪
Both entities are characterized by recurrent crops of spontaneously regressing erythematous papules
- ▪
Lesions of PLEVA are crusted and occasionally vesiculopustular, whereas in PLC they are scaly
- ▪
Individual patients may show intermediate or mixed lesions
- ▪
Histologically, an interface dermatitis with necrotic keratinocytes is seen
- ▪
The infiltrate is predominantly T cells that are often monoclonal
Introduction
The acute and chronic forms of pityriasis lichenoides exist on a disease spectrum with variable presentations that can pose diagnostic and therapeutic challenges . These are papular, often clonal T-cell disorders that may rarely be associated with mycosis fungoides.
History
Pityriasis lichenoides, including its two main variants PLEVA and PLC, was first described between 1894 and 1925. Mucha and Habermann are credited with recognition of PLEVA , while Juliusberg is associated with PLC . Pityriasis lichenoides was included in Brocq’s 1902 treatise on a group of chronic, idiopathic dermatoses that he termed “parapsoriasis”.
Epidemiology
Pityriasis lichenoides is more prevalent in the pediatric population, but it affects patients in all age groups, races, and geographic regions. There is a male predominance.
Pathogenesis
The etiology of pityriasis lichenoides is unknown. It has been postulated to be a response to foreign antigens such as infectious agents and drugs. A few reports describe its association with specific infections (e.g. HIV, parvovirus B19), medications (e.g. estrogen–progesterone, TNF-α inhibitors [infliximab, adalimumab], statins), and radiocontrast dye. Of note, increased numbers of maternally derived keratinocytes have been detected within the epidermis of young male patients with pityriasis lichenoides (as compared to normal skin) and perhaps they triggered a host (child)-versus-graft (mother) disease.
Both PLEVA and PLC contain lesional T-cell infiltrates, with a general predominance of CD8 + cells in PLEVA and CD4 + cells in PLC . Rarely, infiltrates are rich in T cells expressing the γ/δ T-cell receptor . Both types of lesions can exhibit dominant T-cell clonality, although it is more easily demonstrated in PLEVA, where the infiltrate is denser . This clonality indicates that pityriasis lichenoides is a T-cell lymphoproliferative disorder like lymphomatoid papulosis and some forms of T-cell cutaneous lymphoid hyperplasia. Indeed, some patients with lymphomatoid papulosis develop individual lesions indistinguishable from pityriasis lichenoides, suggesting that these two conditions may be related . The concept of pityriasis lichenoides as a T-cell lymphoproliferative disorder may help to explain its occasional association with other lymphoproliferative disorders such as cutaneous T-cell lymphoma, Hodgkin disease, and other lymphomas .
Clinical Features
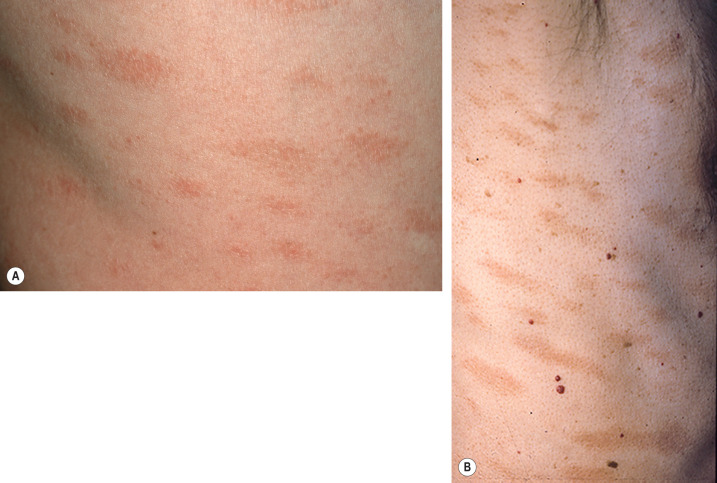
Pityriasis lichenoides presents as recurrent crops of spontaneously regressing erythematous to purpuric papules. The acute form (PLEVA) and the chronic form (PLC) exist on a disease continuum. Many patients have intermediate or mixed manifestations, either serially or concurrently. In patients with PLEVA, individual lesions develop crusts, ulcers, and occasionally vesicles or pustules, which may heal with varioliform scars if dermal damage is extensive ( Fig. 9.3 ). Lesions are usually asymptomatic and typically resolve within weeks. Disease manifestations are confined to the skin, except rarely, when acute lesions are associated with malaise, fever, lymphadenopathy, arthritis, and/or bacteremia. The term febrile ulceronecrotic Mucha–Habermann disease (FUMHD) has been used to refer to such severe variants in which large, confluent necrotic skin lesions as well as mucosal, gastrointestinal, and pulmonary involvement can be observed . Transition from PLEVA to this febrile variant has been associated with increased serum levels of TNF-α.
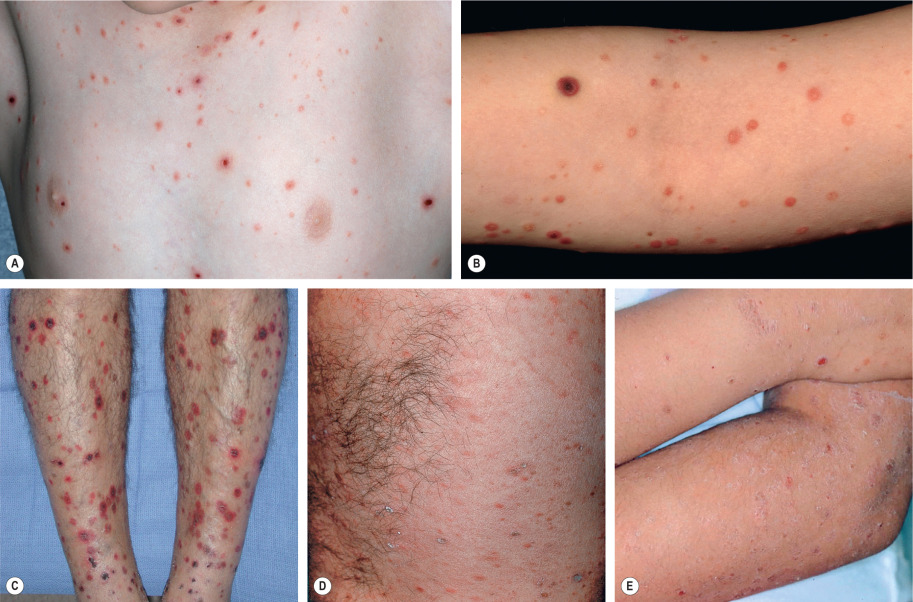
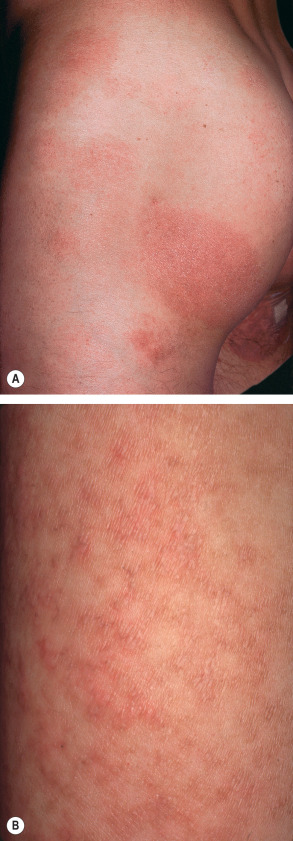
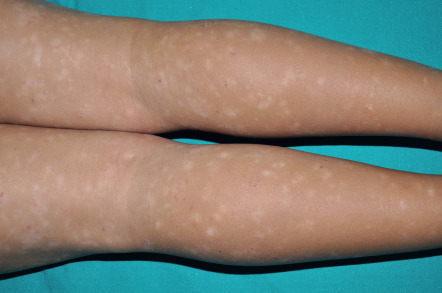
In PLC, the papules are erythematous to red–brown and scaly (see Fig. 9.3 ). They pursue a more indolent course, regressing over weeks to months. When these lesions subside, there may be residual hypopigmented macules; the latter are more obvious in darkly pigmented individuals and may be the presenting complaint ( Fig. 9.4 ). Pityriasis lichenoides can resolve spontaneously after weeks to months or it may pursue a chronic relapsing course, sometimes interspersed with long periods of remission. Some studies have suggested that the distribution of skin lesions is more important than their acute or chronic nature in predicting outcome . Patients with a diffuse distribution of lesions had the shortest average course of disease (11 months), while those with a peripheral distribution had the longest average clinical course (33 months). The central distribution variant was intermediate.
Pathology
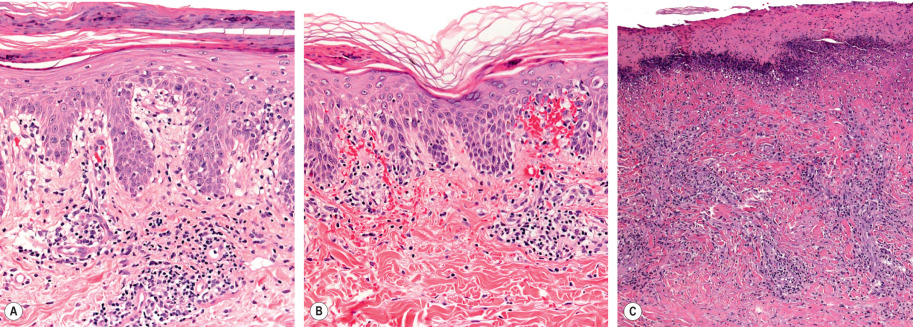
Pityriasis lichenoides exhibits a superficial perivascular interface dermatitis in all cases ( Fig. 9.5 ). Those lesions at the acute end of the spectrum contain a denser infiltrate that may be top-heavy and wedge-shaped. Lymphocytes predominate in the infiltrate, although neutrophils may be admixed. The epidermis shows focal parakeratosis and evidence of damage ranging from edema to extensive epidermal necrosis in well-developed lesions. Extravasation of erythrocytes is a frequent finding. The typical histopathologic alterations accompany the presence of crusts, vesiculopustules, and ulcers. Lymphocytic vasculitis is sometimes described, but true fibrinoid necrosis of blood vessels is not seen (and use of this histologic term can be confusing to nondermatologists). All of these changes are blunted in the more chronic skin lesions, where the principal microscopic features include parakeratosis and a milder interface lymphocytic infiltrate that is accompanied by focal keratinocyte necrosis and mild erythrocyte extravasation. Lymphoid atypia is not a standard feature of pityriasis lichenoides. Although some pathologists allow occasional atypical lymphocytes, others regard this as a sign of lymphomatoid papulosis or another form of CTCL.
Differential Diagnosis
The diagnosis of pityriasis lichenoides is made by the correlation of clinical features with lesional histopathology. The principal differential diagnostic considerations for pityriasis lichenoides are listed in Table 9.3 . For PLEVA, the primary entities in the clinical and histopathologic differential diagnosis include lymphomatoid papulosis, arthropod reactions, cutaneous small vessel vasculitis, varicella, and drug eruptions. Especially in children, mycosis fungoides may histopathologically mimic PLEVA . For PLC, the principal differential diagnosis includes small plaque parapsoriasis, guttate psoriasis, lichen planus (exanthematous), pityriasis rosea, secondary syphilis, lymphomatoid papulosis, drug eruptions, and papular dermatitis. All of these alternative diagnoses can usually be excluded on the basis of key historical, clinical, pathologic, and laboratory findings. More than one biopsy specimen may be required to achieve clinicopathologic correlation.
| PRINCIPAL DIFFERENTIAL DIAGNOSIS OF PITYRIASIS LICHENOIDES ET VARIOLIFORMIS ACUTA (PLEVA) AND PITYRIASIS LICHENOIDES CHRONICA (PLC) | |
|---|---|
| PLEVA | PLC |
| Lymphomatoid papulosis | Small plaque parapsoriasis |
| Cutaneous small vessel vasculitis | Guttate psoriasis |
| Varicella, enteroviral exanthems | Lichen planus (exanthematous) |
| Arthropod reactions | Pityriasis rosea |
| Erythema multiforme | Secondary syphilis |
| Lichenoid drug eruption | Lymphomatoid papulosis |
| Folliculitis | Papular dermatitis |
| Dermatitis herpetiformis | Lichenoid drug eruption |
| When hypopigmented, extrafacial pityriasis alba | |
Aside from immunopathology, most other laboratory tests offer little diagnostic value. A preponderance of CD8 + T cells supports the diagnosis of PLEVA because there are few skin disorders that share this feature. Conversely, the demonstration of large, atypical CD30 + cells generally excludes pityriasis lichenoides and helps to establish the diagnosis of lymphomatoid papulosis. Direct fluorescent antibody (DFA), PCR, and serologic testing as well as immunohistochemical staining are helpful in excluding varicella and syphilis.
Treatment
All treatments for pityriasis lichenoides are based primarily on uncontrolled case series, case reports or anecdotes ( Table 9.4 ). If a drug association is suspected, trial discontinuance of the suspected agent is warranted. First-line therapy includes topical corticosteroids, topical coal tar preparations, tetracyclines, erythromycin, and various types of phototherapy. Oral tetracyclines and erythromycin are used for their anti-inflammatory rather than antibiotic effects, with erythromycin favored in children. A several-month course is often required, followed by a gradual taper. Other antibiotics may be employed if there is secondary infection, usually with Staphylococcus aureus .
More fulminant cases may require low-dose weekly methotrexate. Those rare cases associated with fever and arthritis may benefit from systemic corticosteroids, IVIg or cyclosporine, once infection has been excluded. Antihistamines may be helpful in those instances where significant pruritus is present. There are also reports of the efficacy of TNF-α inhibitors, oral bromelain (bromelin), and photodynamic therapy.
Pityriasis Rubra Pilaris
▪ Lichen ruber pilaris ▪ Devergie disease ▪ Lichen ruber acuminatus
- ▪
Follicular papules with an erythematous base are a characteristic finding, including on the proximal dorsal fingers
- ▪
There is a coalescence of orange–red plaques, but with obvious islands of sparing
- ▪
An orange–red waxy keratoderma of the palms and soles is often seen
- ▪
Of the six types described, the classic adult form is the most common
- ▪
The classic types (adult and juvenile) typically resolve within 3–5 years
- ▪
Histologically, there is alternating ortho- and parakeratosis, both vertically and horizontally
History
Pityriasis rubra pilaris (PRP) was first described in 1835 by Claudius Tarral as a variant of psoriasis. Devergie later recognized it as a separate entity in 1857 and named it “pityriasis pilaris”. In 1889, Besnier recommended the name “pityriasis rubra pilaris”, and it has persisted.
Epidemiology
Although PRP shows no gender bias, affecting both men and women equally, ethnic variations may exist. For example, in one report, the incidence in Great Britain was 1 in 5000 new patient visits whereas in India it was 1 in 50 000 visits. The incidence has either two or three peaks. The first is during the first and second decades and the second is during the sixth decade. The third peak, if real, results from splitting the first and second decades into two separate peaks.
Most cases are acquired, but there are familial forms . Both autosomal dominant and less frequently autosomal recessive inheritance patterns have been described. The dominantly inherited form of PRP is associated with heterozygous gain-of-function mutations in CARD14 , also known as the PSORS2 psoriasis susceptibility gene . Except for the latter group of patients and those with the non-classic forms, PRP appears to be a self-limited disease process, resolving within 3 to 5 years in almost all cases.
Pathogenesis
With the exception of inherited PRP due to CARD14 mutations, no clear etiology has been established. Early on, a vitamin A deficiency was proposed, but this has not been substantiated. The therapeutic success of systemic retinoids suggests a possible dysfunction in keratinization or vitamin A metabolism. Infections, UV exposure, and various minor traumas to the skin have been reported to precede the onset of PRP, implicating a physical trigger or superantigen in selected patients.
The possibility of an autoimmune pathogenesis has also been raised. For example, there are reports of PRP in association with myasthenia gravis, celiac disease, myositis, inflammatory arthritis, and hypothyroidism. An abnormal immunologic response to particular antigens has been suggested by the coexistence of PRP with HIV infection (type VI) , as well as with internal malignancies (e.g. renal cell, bronchogenic and hepatocellular carcinomas). Rarely, drugs have been implicated in triggering PRP, including imatinib, ponatinib, and sofosbuvir .
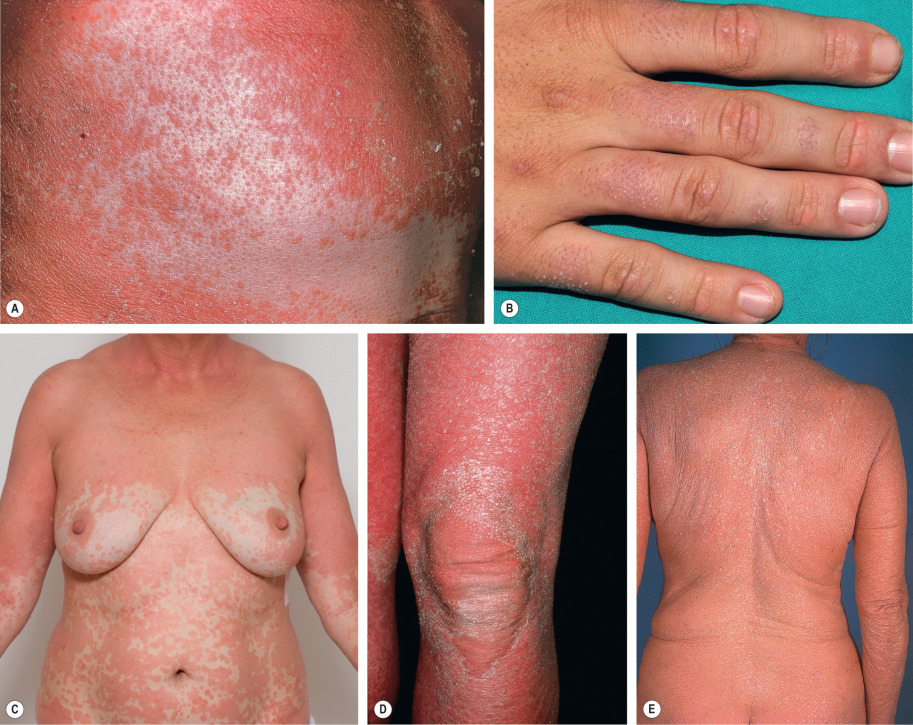
Clinical Features
The multiple, well-described clinical features of classic PRP are helpful in making the diagnosis. Follicular hyperkeratosis on an erythematous base is a key finding ( Fig. 9.6 ). This results in rough papules, especially on the dorsal aspect of the proximal fingers, which have been described as reminiscent of a nutmeg grater. These papules are also seen on the trunk and extremities, and they can coalesce to form large salmon-colored to orange–red plaques with distinctive “islands of sparing”. The plaques can then progress to an erythrodermic appearance with varying degrees of exfoliation (see Ch. 10 ). The palms and soles are commonly involved with a distinctive orange–red waxy keratoderma ( Fig. 9.7 ). Erythema with a fine diffuse scale is often seen on the scalp . Scalp disease that mimics seborrheic dermatitis is a common initial clinical manifestation of adult PRP, and rapid progression to erythroderma can occur over several weeks.