Abstract
In addition to primary and secondary B- and T-cell lymphomas, there are benign lymphocytic infiltrates of the skin that can sometimes mimic cutaneous lymphomas. The latter include lymphocytic infiltrate of Jessner and cutaneous lymphoid hyperplasia (pseudolymphoma). Pseudolymphomas can arise at the site of arthropod bites and tattoos or as reactions to drugs and viral infections or vaccinations. Occasionally, extramedullary hematopoiesis can occur within the skin, and in neonates this presents as a “blueberry muffin baby”. Cutaneous manifestations of hematologic malignancies vary from the pink papules of chronic lymphocytic or acute myelogenous leukemia to the purpuric nodules of blastic plasmacytoid dendritic cell neoplasm. Rarely, leukemia cutis occurs in the absence of circulating leukemic cells. Patients with an underlying leukemia can also develop inflammatory dermatoses such as Sweet syndrome and exaggerated arthropod reactions. Unusual neoplasms, including angioimmunoblastic T-cell lymphoma and lymphomatoid granulomatosis, are also reviewed in this chapter. As in cutaneous lymphomas, clinicopathologic correlation is essential.
Keywords
lymphocytic infiltrate of Jessner, cutaneous lymphoid hyperplasia, pseudolymphoma of the skin, extramedullary hematopoiesis, leukemia cutis, cutaneous Hodgkin lymphoma, blastic plasmacytoid dendritic cell neoplasm, angioimmunoblastic T-cell lymphoma, lymphomatoid granulomatosis, cutaneous plasmacytosis
Benign Lymphocytic Infiltrates
Lymphocytic Infiltrate of Jessner
▪ Lymphocytic infiltrate of Jessner–Kanof ▪ Benign lymphocytic infiltrate of the skin ▪ Jessner lymphocytic infiltrate ofthe skin ▪ Jessner lymphocytic infiltration of the skin ▪ Jessner–Kanof lymphocytic infiltration of the skin
- ▪
Erythematous papules, plaques and, less commonly, nodules
- ▪
Most common on the head, neck and back
- ▪
Dermal infiltrate of lymphocytes without epidermal involvement
History
Lymphocytic infiltrate of Jessner (LIJ) was initially described in 1953 by Jessner and Kanof as circinate papules on the face, surrounding a central area of clearing.
Epidemiology
This entity occurs primarily in middle-aged adults. There is no gender predilection.
Pathogenesis
Benign lymphocytic infiltrate of the skin remains a controversial entity. Some authors believe it is a variant of either lupus erythematosus (primarily LE tumidus) or polymorphic light eruption versus a form of cutaneous lymphoid hyperplasia (pseudolymphoma). There are cases of co-occurrence with LE and with polymorphic light eruption, while in Europe, it has been associated with Borrelia burgdorferi infection . Lastly, rare cases of drug-induced disease (e.g. angiotensin-converting enzyme [ACE] inhibitors, glatiramer acetate) have been described as have familial cases .
Clinical features
Cutaneous lesions appear primarily on the head, neck and upper back as one or several asymptomatic, erythematous papules, plaques, and less commonly nodules, with an absence of secondary epidermal changes such as scale ( Fig. 121.1 ). Annular plaques with central clearing are commonly observed and individual lesions last several weeks to months. There are no associated systemic manifestations. Although spontaneous resolution occurs, recurrences are common.
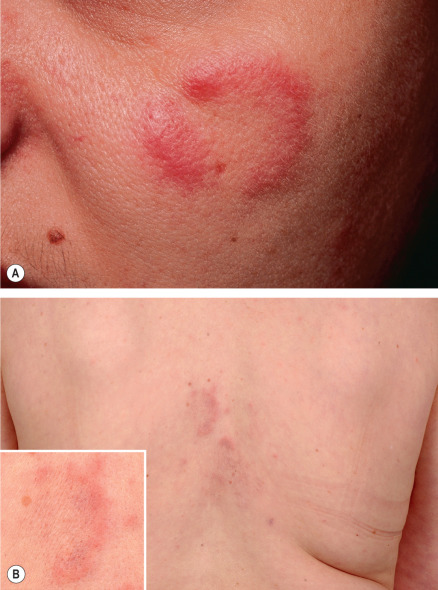
Pathology
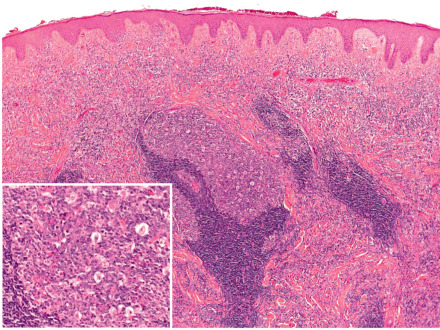
The epidermis is unremarkable, with little evidence of interface dermatitis. There is a superficial and deep, primarily perivascular, lymphocytic infiltrate that may surround hair follicles ( Fig. 121.2 ). A mild increase in dermal mucin may be seen. By immunohistochemistry, a mixed T-cell infiltrate with a predominance of CD8 + lymphocytes is observed , admixed with CD123 + plasmacytoid dendritic cells. The distribution of plasmacytoid dendritic cells is identical to that seen in LE tumidus, further supporting a close relationship .
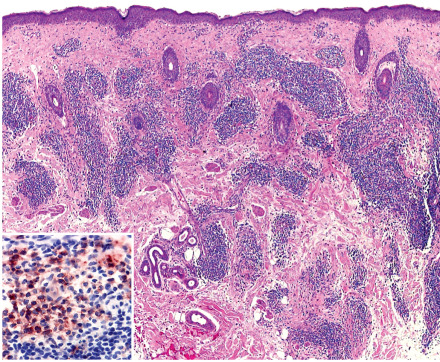
Differential diagnosis
The differential diagnosis includes the plaque form of polymorphic light eruption, LE tumidus, cutaneous lymphoid hyperplasia (pseudolymphoma), reticular erythematous mucinosis, and cutaneous lymphoma ( Fig. 121.3 ). Subacute and chronic cutaneous LE are distinguished by the presence of secondary changes, including scale, follicular plugging and central hypopigmentation, along with interface changes histologically. Because interface changes are sparse, if present at all, in LE tumidus, distinction from LIJ may prove impossible. Reticular erythematous mucinosis, which is considered by some as synonymous with LE tumidus, has abundant dermal mucin.
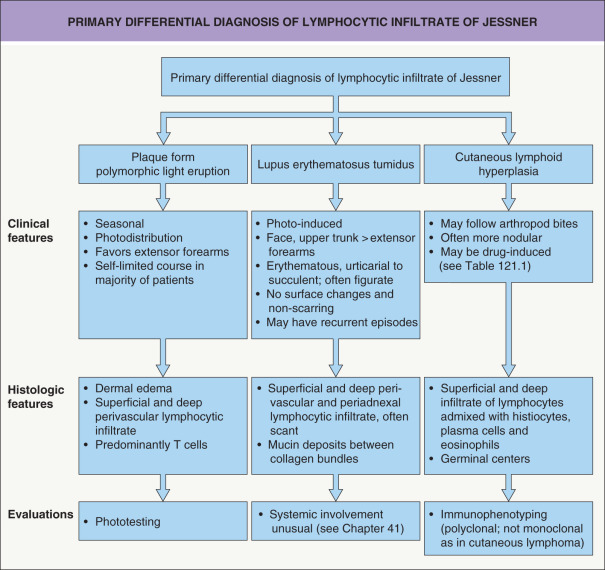
Polymorphic light eruption is associated with sun exposure and usually has a self-limited clinical course. Histologically, papillary dermal edema is often seen and usually there are no clusters of CD123 + plasmacytoid dendritic cells. For some patients, phototesting may provide additional helpful information (see Fig. 121.3 ).
Cutaneous lymphomas can have significant clinical and histopathologic overlap with LIJ. If the dermal lymphocytic infiltrate is extensive, immunophenotypic analyses may help to distinguish between the two entities (see Chs 119 & 120 ). Although individual lesions of borderline lepromatous leprosy may resemble LIJ, these patients have a greater number of widespread lesions.
Treatment
The cutaneous manifestations of LIJ may resolve spontaneously within months to years, without scarring. Oral antibiotics and topical or intralesional corticosteroids have been used with limited success. Up to 50% of patients may improve with hydroxychloroquine. In general, the disorder is resistant to radiation therapy. A crossover study of oral thalidomide (100 mg/day) versus placebo in 25 patients resulted in a clinical response of 76% for thalidomide and 16% for placebo. In isolated case reports, improvement following pulsed dye (595 nm) laser treatment or chemotherapy (for an unrelated malignancy) has been described .
Cutaneous Lymphoid Hyperplasia (Pseudolymphoma of the Skin)
▪ Lymphocytoma cutis ▪ Lymphadenosis benigna cutis(LABC) ▪ Spiegler–Fendt sarcoid ▪ Miliary lymphocytoma
- ▪
Often an isolated, firm, erythematous to violaceous nodule or plaque that measures 1–3 cm in diameter
- ▪
Most common on the face and upper trunk
- ▪
Mixed dermal infiltrate of lymphocytes, plasma cells, and eosinophils; reactive germinal centers
- ▪
Polyclonal inflammatory infiltrate
History
Cutaneous lymphoid hyperplasia (pseudolymphoma) was first reported by Spiegler in 1894. He described patients who presented with clinical features of a malignant neoplasm but experienced a benign clinical course. Although a characteristic feature is the resemblance to cutaneous lymphoma, there is a spectrum of clinical and histologic findings, reflecting its heterogeneous nature .
Epidemiology
Precise data regarding incidence, prevalence, and geographic distribution are lacking. Both children and adults can be affected.
Pathogenesis
Cutaneous lymphoid hyperplasia (CLH) does not represent a single disease, but rather reflects an exaggerated local immunologic reaction to a stimulus, often unrecognized. The possibility of a hapten-driven immunologic response to cells damaged by a direct toxic effect of stimuli has been suggested . Inciting agents include arthropod bites (including ticks and mites), tattoos, metal implants, contact allergens, vaccinations, and medications ( Table 121.1 ) . In addition, several infections such as herpes zoster and Lyme borreliosis have been associated with CLH .
| MEDICATIONS ASSOCIATED WITH CUTANEOUS LYMPHOID HYPERPLASIA (PSEUDOLYMPHOMA) | |
|---|---|
| Anticonvulsants | Carbamazepine , phenobarbital , phenytoin , ethosuximide , lamitrogine , valproic acid , ethotoin, gabapentin |
| Antiarrhythmics | Mexiletine , procainamide |
| Antibiotics | Trimethoprim–sulfamethoxazole , cefixime , cefuroxime, nitrofurantoin , clarithromycin, vancomycin , rifampin , levofloxacin |
| Antidepressants | Amytriptyline , desipramine, fluoxetine, clomipramine, lithium, sertraline |
| Antihistamines | Cimetidine, ranitidine, nizatidine, doxepin |
| Antihypertensives (including diuretics) | Atenolol , captopril , lisinopril, diltiazem , amlodipine, furosemide, amiloride, losartan, valsartan, clonidine patch, metoprolol |
| Antipsychotics | Chlorpromazine, perphenazine, thioridazine |
| Chemotherapeutic agents | Imatinib , fluorouracil, gemcitabine, oxaliplatin, tamoxifen, leucovorin |
| Lipid-lowering agents | Gemfibrozil, lovastatin |
| Nonsteroidal anti-inflammatory drugs (NSAIDs) | Aspirin , ibuprofen , naproxen , diclofenac, indomethacin, ketoprofen, sulindac, diflunisal, oxaprozin, nabumetone |
| Rheumatologic agents | Allopurinol , dapsone , sulfasalazine, TNF inhibitors, methotrexate , gold, D-penicillamine, tocilizumab |
| Sedatives | Benzodiazepines |
| Steroid hormones | Estrogen, progesterone |
| Stimulants | Methylphenidate |
| Vaccines | Hepatitis A, hepatitis B, varicella–zoster |
| Miscellaneous | Cyclosporine, interferon-alpha, aldesleukin (interleukin-2), glatiramer acetate, bromocriptine, black cohosh |
Clinical features
CLH usually presents as a single, 1–3 cm, firm, erythematous to violaceous plaque or nodule located on the head, neck or upper extremities ( Fig. 121.4A,B ). That said, presentations can vary from multiple clustered papules to larger panniculitis-like nodules . The vast majority of lesions lack surface changes such as scale.
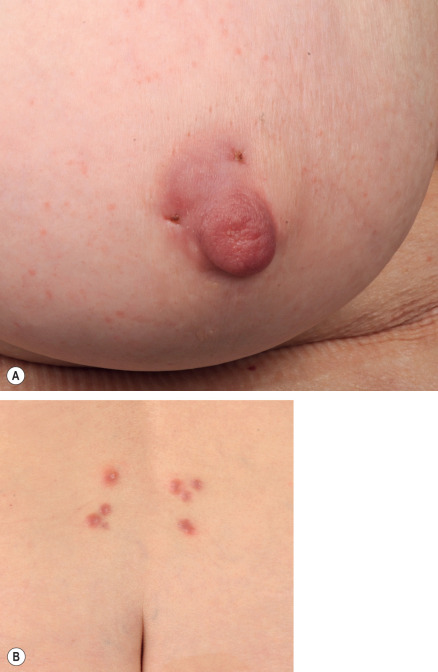
Pathology
The histopathological features are variable and the pattern may resemble either a low-grade B-cell lymphoma (follicle center lymphoma, marginal zone lymphoma) or a T-cell lymphoma, in particular mycosis fungoides, lymphomatoid papulosis (LyP), cutaneous anaplastic large cell lymphoma (cALCL), or subcutaneous T-cell lymphoma .
In CLH with B-cell predominance , there is typically a superficial and deep nodular or diffuse infiltrate of lymphocytes, admixed with histiocytes and occasional plasma cells and eosinophils. In florid cases, germinal centers with prominent tingible-body macrophages may be observed ( Fig. 121.5 ). Distinction from follicle center lymphoma can be made on routine histologic sections or with the addition of immunohistochemical stains ( Table 121.2 ). Clonality, based upon IgH gene rearrangements or restricted κ or λ expression, is usually not seen in pseudolymphomas (see Fig. 119.10 ) . PCR analysis for Borrelia burgdorferi DNA is positive in Borrelia -associated lymphocytoma cutis and pseudolymphomatous acrodermatitis chronica atrophicans .
| MICROSCOPIC DIFFERENCES BETWEEN CUTANEOUS LYMPHOID HYPERPLASIA (PSEUDOLYMPHOMA) AND CUTANEOUS FOLLICLE CENTER B-CELL LYMPHOMA | |
|---|---|
| Cutaneous lymphoid hyperplasia (pseudolymphoma) | Cutaneous follicle center B-cell lymphoma |
| Histologic features | |
| Mixed cellular infiltrate, including eosinophils and plasma cells | Predominantly lymphocytic infiltrate |
| Reactive germinal centers | Neoplastic germinal centers |
| Mantle zone present | No or reduced mantle zone |
| Tingible-body macrophages present | No tingible-body macrophages |
| Polarization of follicles with light and dark areas | Monomorphous appearance of follicles without polarization |
| Immunophenotypic features | |
| T and B lymphocytes | Predominance of B lymphocytes (CD20 + ) |
| Bcl-6 + cells restricted to lymph follicles | Clusters of Bcl-6 + cells outside of lymph follicles |
| Bcl-2 only on T lymphocytes | Bcl-2 on neoplastic B cells (small minority of cases) |
| High proliferation of germinal centers | Reduced proliferation of germinal centers |
| Mixed κ and λ expression | Restricted κ or λ expression |
In T-cell-predominant infiltrates, CD4 + T helper lymphocytes are commonly observed within the dermis, admixed with a minority of CD8 + cytotoxic/suppressor T cells. A mycosis fungoides-like pattern is most often encountered in drug-induced pseudolymphomas (see Table 121.1 ). Histologically, epidermotropism, spongiosis, vacuolar degeneration of the basal layer, papillary dermal edema, and red cell extravasation may be seen. Prominent papillary dermal fibrosis, which can be seen in mycosis fungoides, is absent, but T-cell clonality may be present.
CD30 + pseudolymphomas include persistent arthropod bite reactions, drug reactions, and the atypical lymphoid infiltrates associated with cutaneous poxvirus infections (e.g. molluscum contagiosum, orf). Additional findings related to the underlying disorder can aid in diagnosis, and there is polyclonal rearrangement of the TCR .
Differential diagnosis
Pseudolymphomas may be difficult to differentiate from cutaneous lymphomas (see Pathology section). Although the presence of clonality may aid in distinguishing reactive from neoplastic processes, it should not be used to establish malignancy. In addition to clinicopathologic correlation, longitudinal evaluation plays a key role in making this distinction.
Acral pseudolymphomatous angiokeratoma of children (APACHE), originally thought to represent a vascular nevus, is now categorized as a pseudolymphoma. In contrast to other pseudolymphomas, it favors the extremities of children between the ages of 2 and 16 years, usually presenting as a unilateral grouping of small, red to violet, “angiomatous” papules. Histologically, there is a dermal infiltrate of lymphocytes, histiocytes and plasma cells accompanied by prominently thickened capillaries .
Kikuchi–Fujimoto disease , also known as histiocytic necrotizing lymphadenitis, is an idiopathic systemic inflammatory disease most commonly seen in young adult women. It is associated with a variety of viral and bacterial infections (e.g. Epstein–Barr virus, cytomegalovirus) as well as autoimmune diseases (e.g. systemic LE). In addition to fevers, malaise, weight loss and gastrointestinal symptoms, patients develop cervical lymphadenopathy. Histologically, there is necrosis and histiocytic infiltration of the paracortical areas of involved lymph nodes, without an identifiable pathogen. Cutaneous findings, present in 40% of patients, include acneiform eruptions, urticaria, ulcers and indurated erythematous plaques. Histologically, dense superficial and deep perivascular lymphohistiocytic infiltrates with nuclear debris (in the absence of neutrophils) are seen; interface changes are commonly present .
In “pseudolymphomatous folliculitis” , there is typically a solitary facial nodule that is due to a predominantly perifollicular and periadnexal dermal infiltrate. The latter is typically mixed, with a predominance of T or B cells that may have cellular atypia; granulomas are variably present . There is irregular hyperplasia and distortion of the follicular epithelium, with blurring of the dermal–epidermal junction, and infiltrates of CD1a + /S100 + mononuclear cells surround and infiltrate these distorted follicles. This entity should be distinguished from primary cutaneous CD4 + small/medium-sized pleomorphic T-cell lymphoproliferative disorder, which is characterized by more pronounced pleomorphism and often has phenotypic aberrations such as PD1 expression .
Cutaneous and systemic plasmacytosis is a benign condition of unknown etiology observed primarily in Asians, especially those of Japanese ancestry. It is characterized by multiple, reddish-brown, infiltrated maculopapules and plaques, most often on the trunk. Histopathologically, variably dense superficial and deep perivascular infiltrates composed predominantly of mature polyclonal plasma cells are seen . The plasmacytosis may be accompanied by anemia, B symptoms and hypergammaglobulinemia, as well as lymphadenopathy, hepatosplenomegaly, interstitial pneumonia, and mesangial proliferative glomerulonephritis. A reactive dysfunction of plasma cells triggered by various stimuli has been postulated . As in Castleman disease, there is an elevation in circulating/tissue IL-6, but in the absence of HHV-8 infection .
Additional examples of pseudolymphoma are outlined in Table 121.3 .
| ENTITIES THAT CAN MIMIC LYMPHOMA CUTIS (PSEUDOLYMPHOMAS) | ||
|---|---|---|
| More common | Uncommon | Rare |
| Persistent arthropod bites, scabies | Lichen sclerosus on genital skin Lymphomatoid contact dermatitis Pigmented purpura, lichenoid variant Pityriasis lichenoides et varioliformis acuta (PLEVA), especially ulceronecrotic Pseudolymphomatous atopic dermatitis Pseudolymphomatous folliculitis Pseudolymphomatous reactions to tattoo pigments Syphilis, primary and secondary | Actinic reticuloid APACHE (acral pseudolymphomatous angiokeratoma of children) Cutaneous IgG4-related disease * Cutaneous plasmacytosis Hydroa vacciniforme Intralymphatic histiocytosis Kikuchi–Fujimoto disease Lobular panniculitis in children with congenital immunodeficiencies Pseudolymphomatous reactions to vaccinations Vitiligo (inflammatory stage) |
| Borrelia infections, in particular lymphocytoma cutis and acrodermatitis chronica atrophicans (Europe) | ||
| CD30 + reactive lymphoid infiltrates in the context of cutaneous infections (e.g. herpes simplex, herpes zoster, molluscum contagiosum) | ||
| Drug eruptions (lichenoid, lymphomatoid) (see Table 121.1 ) | ||
| Lichenoid (lymphomatoid) keratosis | ||
| Pseudolymphomatous infiltrates in lupus erythematosus | ||
* Increased IgG4-positive plasma calls and sclerotic stroma; can also see granuloma faciale-like lesions.
Treatment
CLH is a benign, reactive condition and it should be treated conservatively. Spontaneous resolution may occur without scarring. For persistent lesions, topical and/or intralesional corticosteroids may lead to improvement. Simple excision, cryosurgery, laser ablation, and radiation therapy are other options. Thalidomide has been utilized with success in recalcitrant cases . Performance of a biopsy, even if partial, can be followed by spontaneous regression. For Borrelia -associated pseudolymphoma, appropriate antibiotics are administered (see Ch. 19 ).
Extramedullary Hematopoiesis
- ▪
Evidence of bone marrow dysfunction
- ▪
Most common associations are TORCH ( t oxoplasmosis, o ther agents, r ubella, c ytomegalovirus and h erpes simplex virus) infections in neonates and myelofibrosis in adults
- ▪
Widely disseminated erythematous to violaceous papules and nodules, with neonatal form referred to as “blueberry muffi baby”
- ▪
Diffuse dermal infiltrate of immature erythrocytes, leukocytes and megakaryocytes
Epidemiology
Extramedullary hematopoiesis (EMH) is a reflection of bone marrow dysfunction and is most commonly seen in neonates. It may occur in adults with myelofibrosis and less often myelodysplasia or after splenectomy .
Pathogenesis
Cutaneous EMH occurs normally during early embryogenesis and abates prior to birth. Thereafter, it occurs only as a secondary phenomenon in response to altered bone marrow function. Rarely, in otherwise healthy individuals, primary cutaneous neoplasms such as pilomatricomas, nevus sebaceus, hemangiomas, and pyogenic granulomas may be associated with localized EMH .
Clinical Features
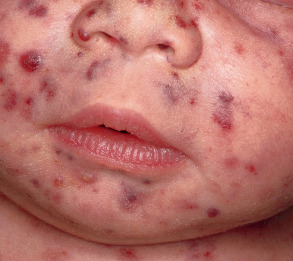
The clinical presentation consists of erythematous to violaceous papules and nodules which may ulcerate. When widely disseminated in neonates, it leads to the classic “blueberry muffin baby” ( Fig. 121.6 ) . This can be seen in association with congenital viral infections and prenatal anemias ( Table 121.4 ). Dermal hematopoiesis is most often seen with rubella and cytomegalovirus, and serologic titers for TORCH infections can aid in diagnosis.
| DIFFERENTIAL DIAGNOSIS OF “BLUEBERRY MUFFIN BABY” |
Disseminated extramedullary hematopoiesis
Congenital leukemia cutis Neonatal neuroblastoma Congenital Langerhans cell histiocytosis Congenital alveolar rhabdomyosarcoma Hemangiomatosis, other vascular lesions (e.g. multifocal lymphangioendotheliomatosis, glomuvenous malformations) |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


