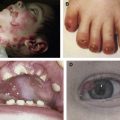
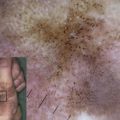
Non-Herlitz junctional epidermolysis bullosa (nH JEB) is characterized by generalized blisters that predominate in sites exposed to friction, trauma, or heat. Whereas infants and children with nH JEB often appear to resemble patients with other forms of EB, adults with this disorder typically display atrophic scars, hypopigmentation, or hyperpigmentation at sites of healed blisters as well as incomplete alopecia, dystrophic nails, mucous membrane involvement, and dental abnormalities. Mild (or severe) disease early in life may be characterized by the opposite phenotype in adults with nH JEB. Although nH JEB is generally less severe than Herlitz disease, fatalities (especially in neonates) are not uncommon among patients with the former diagnosis.
Epidermolysis bullosa (EB) is the term applied to a heterogeneous group of inherited blistering diseases in which minor trauma leads to blistering of skin and mucous membranes. Three major groups of EB have been defined according to the level of blister formation that occurs in epidermal basement membrane (BM): (1) EB simplex, in which blisters form within basal keratinocytes; (2) junctional EB (JEB), in which blisters form within the lamina lucida of the epidermal BM; and (3) dystrophic EB, in which blisters form below the lamina densa. These 3 major groups are further divided into several subtypes based on their pattern of inheritance, morphology of lesions, or the distribution of involvement. The severity of these diseases ranges from mild, with almost no functional impairment, to devastating, with life-threatening complications and fatal outcomes. Junctional EB (JEB) represents a group of disorders characterized by autosomal recessive inheritance (though a rare exception has recently been reported ) and manifestation of disease activity at birth (in almost all subtypes). The various types of JEB are summarized in Table 1 . Most EB classification schemes place emphasis on distinguishing Herlitz from non-Herlitz forms of JEB, because the former typically has a far less favorable prognosis (hence its prior designation as EB lethalis or the gravis variant of generalized JEB). Several variant forms of non-Herlitz JEB (nH JEB) are currently recognized (see Table 1 ). This review focuses on nH JEB generalized, nH JEB localized, nH JEB inversa, and nH JEB late onset (EB progressive), as entities currently classified as other JEB variants are detailed elsewhere in this monograph.
| Subtype | Disease Gene | Protein Target |
|---|---|---|
| H JEB | LAMA3 , LAMB3 , LAMC2 | Laminin 332 |
| nH JEB | COL17A1 | Collagen XVII |
| LAMA3 , LAMB3 , LAMC2 | Laminin 332 | |
| nH JEB, localized | COL17A1 | Collagen XVII |
| JEB, inversa | LAMA3 , LAMB3 , LAMC2 | Laminin 332 |
| JEB, progressiva | ? | ? |
| JEB with pyloric atresia | ITGA6 , ITGB4 | Integrin subunits α 6 and β 4 respectively |
History
In 1935, Herlitz described a form of EB that was lethal in infancy. When the skin of patients with this form of EB was evaluated by electron microscopy, a subepidermal blister was evident within the plane of the lamina lucida. Early on, therefore, JEB was considered synonymous with Herlitz disease. The grim prognosis for this disease made it remarkable when, in 1976, Hashimoto and colleagues described a patient with JEB who survived to adulthood. The proband was a 38 year-old man who displayed generalized blistering of skin and mucous membranes since birth, atrophy in areas of repeated blistering, alopecia, dystrophic nails, loss of teeth, and blister formation within the lamina lucida. Of note, this patient had 2 siblings who died in their first days of life due to blistering and associated complications. Hashimoto named this form of JEB the Disentis type, based on the place of birth of these Swiss patients. Four more patients with similar clinical and electron microscopic features were subsequently described in 1979 by Anton-Lamprecht and Schnyder. The dramatically different prognosis in these patients prompted a new appellation for this subtype of JEB, namely EB atrophicans generalisata mitis. In 1982, Hintner and Wolff described 8 additional patients with nonlethal JEB and anglicized the previous term to generalized atrophic benign epidermolysis bullosa (GABEB). More patients were subsequently described, including additional European cases as well as patients from the United States, Asia, and Northern Africa. Accordingly, nH JEB is now recognized as a sporadic disease that occurs worldwide.
Laboratory findings
Light Microscopy
Light microscopy studies of early lesional skin from patients with nH JEB typically show subepidermal blisters with no signs of inflammation. Induced blisters have the same histologic appearance as early trauma-induced blisters. In periodic acid Schiff (PAS)-stained sections, the BM is found on the blister floor, while intact keratinocytes form the roof. Although light microscopy studies are helpful in the evaluation of these patients, more specialized techniques are currently used to establish the plane of blister formation in these patients’ skin.
Transmission Electron Microscopy
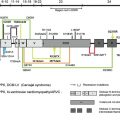
Transmission electron microscopy of skin from patients with nH JEB identifies a blister cleavage plane within the electron lucent lamina lucida subregion of epidermal BM. Whereas immunofluorescence mapping studies can establish this finding, transmission electron microscopy studies offer the potential to identify other relevant findings in the skin of nH JEB patients, namely, a potential reduction in the number or size of hemidesmosomes in epidermal BM ( Table 2 ). However, it should be kept in mind that patients with nH JEB may show a spectrum of ultrastructural findings in their skin; findings perhaps dependent on corresponding mutations in disease genes, patient age, regional variations of skin samples, degree of disease activity in the skin sample under study, or other variables yet to be defined. Transmission electron microscopy of skin from patients with EB is a highly specialized investigation best performed in an experienced reference laboratory (several are enumerated by Fine and colleagues).
| JEB Type | TEM Cleavage Plane | TEM, Other Observations | IF Mapping Studies | Alterations in Structural Proteins |
|---|---|---|---|---|
| H JEB | Lamina lucida | Decreased or absent HDs and/or SBDPs | BPAG1 roof; collagens IV and VII, base | Laminin 332 absent (or markedly decreased) |
| nH JEB | Lamina lucida | HDs within normal limits, or decreased in size and number | BPAG1, roof; collagens IV and VII, base | Type XVII collagen absent (or notably decreased) Laminin 332 attenuated or markedly decreased |
| JEB with pyloric atresia | Lamina lucida | Small plaques in HDs; attenuated SBDPs common | BPAG1 roof; collagens IV and VII, base | Integrin α 6 β 4 absent or markedly decreased |
Immunofluorescence Mapping Studies
The level of blister formation in skin from EB patients can also be defined by immunofluorescence (IF) mapping studies. These studies typically use cryopreserved skin harvested (best by shave/saucerization techniques) from fresh spontaneous or friction-induced blisters. Using monoclonal antibodies directed against reference landmark adhesion proteins that reside in epidermal BM (eg, bullous pemphigoid antigen 1 [BPAG1], type IV collagen, type VII collagen, and so forth), it is usually possible to determine with accuracy whether the plane of cleavage in lesional skin resides within basal keratinocytes (all markers on floor of blister), the lamina lucida (BPAG1 on roof of blister, collagen IV and VII on floor of blister), or the sublamina densa region (all markers on roof of blister). IF mapping studies offer the additional advantage in that inclusion of additional monoclonal antibodies directed against candidate “disease-proteins” (eg, monoclonal antibodies vs type XVII collagen [often altered in patients with nH JEB], monoclonal antibodies vs type VII collagen [altered in patients with dystrophic EB]) has the potential to identify defects in the relative expression or distribution of adhesion proteins within the epidermal BM of patient skin. Indeed, identifying a candidate disease protein (and hence, its corresponding disease gene) greatly facilitates DNA mutational analyses. Prior studies have demonstrated that IF mapping studies are as reliable diagnostically as transmission electron microscopy for the classification of patients with EB. Moreover, there are numerous university-based and commercial laboratories that can perform this testing. A summary overview of what IF mapping studies typically show in nH JEB patients is shown in Table 2 .
Mutational Analysis
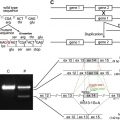
The ultimate means of determining precise defects in patients with EB resides in mutational analysis of genes encoding epidermal BM-associated structural proteins. In addition, identification of mutations responsible for disease in a given patient also has relevance to defining: (a) the exact mode of disease inheritance; (b) what defects require correction (or modulation) by gene therapy; and (c) data sets required for prenatal or preimplantation diagnostics. As noted earlier, the clinical and genetic heterogeneity of EB (especially junctional forms of EB) coupled with the labor-intensive character of mutational analyses places great emphasis on IF mapping and screening studies to narrow candidates for genetic testing. At present, several research laboratories and commercial vendors offer mutational analysis for EB patients.
In JEB, the majority of mutations identified to date in both Herlitz and non-Herlitz forms of disease have resided in the 3 genes, LAMA3 , LAMB3 , and LAMC2 , that encode the α, β, and γ subunits, respectively, of laminin 332. Most mutations in patients with H JEB are nonsense mutations in LAMA3 , LAMB3 , or LAMC2 that result in the formation of premature termination codons (PTC). In turn, such PTCs elicit nonsense-mediated mRNA decay or synthesis of truncated (or nonfunctional) laminin subunit polypeptides. The correlate observation in IF mapping studies of skin from patients with H JEB is complete (or near complete) absence of laminin 332 expression in epidermal BM.
In nH JEB, patients may demonstrate mutations in COL17A1 , the gene encoding type XVII collagen (also termed bullous pemphigoid antigen 2 [BPAG2] or BP180), or the genes encoding laminin 332. In the subset of nH JEB patients with COL17A1 defects, mutations have traditionally been shown to consist of insertions, deletions, or nonsense mutations that result in PTCs, nonsense-mediated mRNA decay, and complete absence of collagen XVII expression in epidermal BM. However, exceptions to this paradigm exist, as evidenced by one patient who carried one COL17A1 allele harboring a dominantly inherited mutation encoding a glycine substitution and one COL17A1 allele bearing a mutation encoding a PTC. In nH JEB patients with laminin 332 gene mutations, it is not uncommon for patients to carry a PTC on one allele and a missense or in-frame splice site mutation on the alternate allele. Such alterations have been thought to result in production of small amounts of truncated or altered subunit polypeptides that retain the ability to be incorporated with laminin 332 heterotrimers within hemidesmosome-anchoring filament complexes. This interpretation is consistent with IF mapping studies that show diminished expression of laminin 332 within the epidermal BM of patients with nH JEB. In nH JEB (and H JEB as well), the majority of the mutations identified to date reside within LAMB3 .
A recent study examining mutations in 265 patients carrying a preliminary diagnosis of JEB generally confirmed the summary observations outlined herein along with several unexpected findings. For example, several patients harboring mutations causing PTCs in genes encoding laminin 332 displayed a relatively mild phenotype that contrasted notably with that traditionally seen in patients with H JEB. In addition, this study identified several patients with an H JEB phenotype that carried no mutations in LAMA3 , LAMB3 , or LAMC2 but instead harbored PTC causing mutations in COL17A1 . These patients not only had disease characterized by greater relative severity but also an increased risk of lethality. Additional summary information about genetic alterations in patients with nH JEB is presented in Table 3 .
| Gene | Types of Mutations | Corresponding Protein |
|---|---|---|
| COL17A1 | Nonsense, missense, insertion, deletion, or splice site mutations that often result in PTCs on both COL17A1 alleles | Type XVII collagen (bullous pemphigoid antigen 2 [BPAG2], BP180) |
| LAMA3 LAMB3 LAMC2 | Nonsense, missense, insertion, deletion, or splice site mutations that often result in a PTC on one allele and missense mutations or in frame insertions/deletions on the other allele | Laminin 332 (also termed laminin 5, epiligrin, nicein, kalinin, or the BM600 antigen) |
Laboratory findings
Light Microscopy
Light microscopy studies of early lesional skin from patients with nH JEB typically show subepidermal blisters with no signs of inflammation. Induced blisters have the same histologic appearance as early trauma-induced blisters. In periodic acid Schiff (PAS)-stained sections, the BM is found on the blister floor, while intact keratinocytes form the roof. Although light microscopy studies are helpful in the evaluation of these patients, more specialized techniques are currently used to establish the plane of blister formation in these patients’ skin.
Transmission Electron Microscopy
Transmission electron microscopy of skin from patients with nH JEB identifies a blister cleavage plane within the electron lucent lamina lucida subregion of epidermal BM. Whereas immunofluorescence mapping studies can establish this finding, transmission electron microscopy studies offer the potential to identify other relevant findings in the skin of nH JEB patients, namely, a potential reduction in the number or size of hemidesmosomes in epidermal BM ( Table 2 ). However, it should be kept in mind that patients with nH JEB may show a spectrum of ultrastructural findings in their skin; findings perhaps dependent on corresponding mutations in disease genes, patient age, regional variations of skin samples, degree of disease activity in the skin sample under study, or other variables yet to be defined. Transmission electron microscopy of skin from patients with EB is a highly specialized investigation best performed in an experienced reference laboratory (several are enumerated by Fine and colleagues).
| JEB Type | TEM Cleavage Plane | TEM, Other Observations | IF Mapping Studies | Alterations in Structural Proteins |
|---|---|---|---|---|
| H JEB | Lamina lucida | Decreased or absent HDs and/or SBDPs | BPAG1 roof; collagens IV and VII, base | Laminin 332 absent (or markedly decreased) |
| nH JEB | Lamina lucida | HDs within normal limits, or decreased in size and number | BPAG1, roof; collagens IV and VII, base | Type XVII collagen absent (or notably decreased) Laminin 332 attenuated or markedly decreased |
| JEB with pyloric atresia | Lamina lucida | Small plaques in HDs; attenuated SBDPs common | BPAG1 roof; collagens IV and VII, base | Integrin α 6 β 4 absent or markedly decreased |
Immunofluorescence Mapping Studies
The level of blister formation in skin from EB patients can also be defined by immunofluorescence (IF) mapping studies. These studies typically use cryopreserved skin harvested (best by shave/saucerization techniques) from fresh spontaneous or friction-induced blisters. Using monoclonal antibodies directed against reference landmark adhesion proteins that reside in epidermal BM (eg, bullous pemphigoid antigen 1 [BPAG1], type IV collagen, type VII collagen, and so forth), it is usually possible to determine with accuracy whether the plane of cleavage in lesional skin resides within basal keratinocytes (all markers on floor of blister), the lamina lucida (BPAG1 on roof of blister, collagen IV and VII on floor of blister), or the sublamina densa region (all markers on roof of blister). IF mapping studies offer the additional advantage in that inclusion of additional monoclonal antibodies directed against candidate “disease-proteins” (eg, monoclonal antibodies vs type XVII collagen [often altered in patients with nH JEB], monoclonal antibodies vs type VII collagen [altered in patients with dystrophic EB]) has the potential to identify defects in the relative expression or distribution of adhesion proteins within the epidermal BM of patient skin. Indeed, identifying a candidate disease protein (and hence, its corresponding disease gene) greatly facilitates DNA mutational analyses. Prior studies have demonstrated that IF mapping studies are as reliable diagnostically as transmission electron microscopy for the classification of patients with EB. Moreover, there are numerous university-based and commercial laboratories that can perform this testing. A summary overview of what IF mapping studies typically show in nH JEB patients is shown in Table 2 .
Mutational Analysis
The ultimate means of determining precise defects in patients with EB resides in mutational analysis of genes encoding epidermal BM-associated structural proteins. In addition, identification of mutations responsible for disease in a given patient also has relevance to defining: (a) the exact mode of disease inheritance; (b) what defects require correction (or modulation) by gene therapy; and (c) data sets required for prenatal or preimplantation diagnostics. As noted earlier, the clinical and genetic heterogeneity of EB (especially junctional forms of EB) coupled with the labor-intensive character of mutational analyses places great emphasis on IF mapping and screening studies to narrow candidates for genetic testing. At present, several research laboratories and commercial vendors offer mutational analysis for EB patients.
In JEB, the majority of mutations identified to date in both Herlitz and non-Herlitz forms of disease have resided in the 3 genes, LAMA3 , LAMB3 , and LAMC2 , that encode the α, β, and γ subunits, respectively, of laminin 332. Most mutations in patients with H JEB are nonsense mutations in LAMA3 , LAMB3 , or LAMC2 that result in the formation of premature termination codons (PTC). In turn, such PTCs elicit nonsense-mediated mRNA decay or synthesis of truncated (or nonfunctional) laminin subunit polypeptides. The correlate observation in IF mapping studies of skin from patients with H JEB is complete (or near complete) absence of laminin 332 expression in epidermal BM.
In nH JEB, patients may demonstrate mutations in COL17A1 , the gene encoding type XVII collagen (also termed bullous pemphigoid antigen 2 [BPAG2] or BP180), or the genes encoding laminin 332. In the subset of nH JEB patients with COL17A1 defects, mutations have traditionally been shown to consist of insertions, deletions, or nonsense mutations that result in PTCs, nonsense-mediated mRNA decay, and complete absence of collagen XVII expression in epidermal BM. However, exceptions to this paradigm exist, as evidenced by one patient who carried one COL17A1 allele harboring a dominantly inherited mutation encoding a glycine substitution and one COL17A1 allele bearing a mutation encoding a PTC. In nH JEB patients with laminin 332 gene mutations, it is not uncommon for patients to carry a PTC on one allele and a missense or in-frame splice site mutation on the alternate allele. Such alterations have been thought to result in production of small amounts of truncated or altered subunit polypeptides that retain the ability to be incorporated with laminin 332 heterotrimers within hemidesmosome-anchoring filament complexes. This interpretation is consistent with IF mapping studies that show diminished expression of laminin 332 within the epidermal BM of patients with nH JEB. In nH JEB (and H JEB as well), the majority of the mutations identified to date reside within LAMB3 .
A recent study examining mutations in 265 patients carrying a preliminary diagnosis of JEB generally confirmed the summary observations outlined herein along with several unexpected findings. For example, several patients harboring mutations causing PTCs in genes encoding laminin 332 displayed a relatively mild phenotype that contrasted notably with that traditionally seen in patients with H JEB. In addition, this study identified several patients with an H JEB phenotype that carried no mutations in LAMA3 , LAMB3 , or LAMC2 but instead harbored PTC causing mutations in COL17A1 . These patients not only had disease characterized by greater relative severity but also an increased risk of lethality. Additional summary information about genetic alterations in patients with nH JEB is presented in Table 3 .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


