Neurofibromas
Lauren L. Levy
Amanda Zubek
Macrene Alexiades
BACKGROUND
Neurofibromas (NFs) are benign nerve sheath tumors. Solitary cutaneous neurofibromas are commonly found in adults and are usually not associated with any systemic or genetic disease. Plexiform neurofibromas occur in up to 30% of cases of neurofibromatosis type 1 (NF1), most frequently in the craniomaxillofacial region. These lesions manifest early in life and are at high risk for transformation into malignant peripheral nerve sheath tumors (MPNSTs).1 Malignant progression is generally considered the main cause of mortality, occurring in 2% to 16% of cases.1 NF1 is a genetic disorder characterized by café-au-lait spots, axillary and inguinal freckling, skeletal dysplasias, both benign and malignant nervous system tumors, and numerous benign neurofibromas.
PRESENTATION
Solitary Neurofibroma
Patients present in adulthood with a complaint of painless cutaneous papulonodules.
Plexiform Neurofibroma
Plexiform neurofibromas present as large, ill-defined subcutaneous swellings. They can range in size from a few centimeters in diameter to those involving a large area of the body. They are occasionally described as having a “bag of worms” texture. The patient usually presents owing to pain at the lesions. Other primary symptoms are ocular motility disturbances from eye involvement, dyspnea and respiratory failure from upper airway compression, neurological deficits from cranial
nerve compression, and social deprivation, depression, and other mood disorders resulting from facial disfigurement.2
nerve compression, and social deprivation, depression, and other mood disorders resulting from facial disfigurement.2
Different clinical presentations can be observed, depending on the location of the tumor and the originating nerve. Plexiform NFs seen on skin examination may involve the skin only and may also represent the surface of a much deeper tumor. These tumors often undergo a rapid growth phase in the early years of life, after which they may remain quiescent, growing only in proportion to body growth; they may continue to grow steadily; or they may have a second peak of rapid growth during puberty or pregnancy in women.2 Some plexiform NFs may impinge on critical structures such as the airway, urinary tract, or spinal nerve roots and thereby cause significant morbidity.
Neurofibromatosis 1
Signs and symptoms of NF1 vary widely. The initial finding observed in children with NF1 is multiple café-au-lait macules (CALMs). Present at birth or appearing over time, CALMS typically increase in size and number throughout childhood. Axillary or inguinal freckles are rarely observed at birth; they develop from childhood through adolescence. Subcutaneous or cutaneous neurofibromas usually appear in older children, adolescents, and adults.3
Additional signs and symptoms may include:
Hypertension (potentially from renal artery stenosis or pheochromocytoma)
Bone abnormalities (congenital pseudoarthrosis, sphenoid bone dysplasia, scoliosis)
Visual abnormalities (optic nerve tumors)
Lisch nodules (iris hamartomas)
Learning disabilities, attention deficit hyperactivity disorder, autism spectrum disorder
Larger-than-average head size (macrocephaly)
Short stature
Neurofibromatosis 2
Patients with NF2 typically present with hearing loss, tinnitus, and balance problems due to vestibular nerve lesions. Cutaneous neurofibromas and schwannomas present as cutaneous or subcutaneous nodules in NF2 and can precede the onset of other signs and symptoms. Patients with NF2 have fewer CALMs than do patients NF1, and axillary and inguinal freckling are absent.3
DIAGNOSIS
Clinical Diagnosis
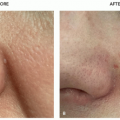
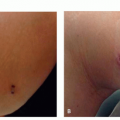
Neurofibromas are soft skin-colored to pink fleshy papules that “buttonhole” when downward pressure is placed on the lesion. Figure 11.8.1 illustrates a solitary neurofibroma. When multiple neurofibromas are present, neurofibromatosis should be considered.
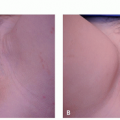
Plexiform neurofibromas are large baggy subcutaneous masses measuring several centimeters in diameter and are considered pathognomonic for NF1, although they can rarely be seen sporadically. Their consistency is usually soft; however, hypertrophic nerve trunks can sometimes be palpated. The overlying skin may display hypertrophy, hyperpigmentation, and hypertrichosis.
Different clinical presentations can be observed, depending on the location of the plexiform NF. In the nodular variant, the nerve trunks develop multiple nodular neurofibromas that merge into one another and may involve a single nerve or a plexus of nerves. The patient usually presents owing to pain at the lesions. When involving superficial nerve trunks, the lesions present as smooth subcutaneous masses, tender to palpation.2 When deeper nerve trunks are involved, the lesions may be detectable only on imaging. Magnetic resonance imaging (MRI) scan should be considered to delineate the extent of internal involvement, and positron emission tomography (PET) imaging may be considered to evaluate for MPNST.4
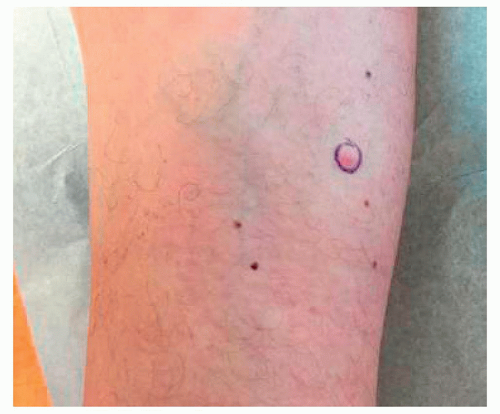 FIGURE 11.8.1 Solitary neurofibroma on the leg of a healthy patient. |
Neurofibromatosis 1
Clinical diagnosis requires the presence of at least 2 of 7 criteria to confirm the presence of NF1. Many of these signs do not appear until later childhood or adolescence; thus, confirming the diagnosis often is delayed despite a suspicion of NF1. The 7 clinical criteria used to diagnose NF1 are as follows, in the absence of alternative diagnoses:
Six or more café-au-lait spots or hyperpigmented macules (at least 5 mm in diameter in prepubertal children and 15 mm postpubertal)
Axillary or inguinal freckles (>2 freckles)
Two or more typical neurofibromas or one plexiform neurofibroma
Optic nerve glioma
Two or more iris hamartomas (Lisch nodules), identified through ophthalmologic slit-lamp examination
Sphenoid dysplasia or typical long-bone abnormalities such as pseudarthrosis
First-degree relative (eg, mother, father, sister, brother) with NF1
Neurofibromatosis 2
Clinical diagnosis of NF2 requires at least 1 of the following 3 constellations of clinical findings:
Bilateral vestibular schwannomas
First-degree relative with NF2 and
Unilateral vestibular schwannoma or
Any 2 of meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
Unilateral vestibular schwannoma and
Any 2 of meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
Multiple meningiomas and
Unilateral vestibular schwannoma or
Any 2 of schwannoma, glioma, neurofibroma, cataract
Histopathology
In a typical neurofibroma, a nonencapsulated lesion is observed in the dermis, representing a proliferation of all elements of peripheral nerves. The cells display wavy serpentine nuclei and pointed ends. Stromal mucin deposition, fibroplasia, and mast cells are characteristic. Axons are present in the tumor, which may be revealed by immunohistochemical stains for neurofilament.1
Plexiform neurofibroma is a subtype in which the NF tumor is encapsulated within a nerve bundle causing the nerve fascicles to expand in an irregular fashion. Although large plexiform neurofibromas are considered diagnostic of NF1, a plexiform pattern may be seen in solitary skin lesions that are not associated with other findings.1,2
In NF2, cutaneous lesions are most often schwannomas (also termed neurilemmomas) on histology. Schwannomas are encapsulated and well circumscribed, with areas of hypercellularity and areas of hypocellularity. In the hypercellular areas, Verocay bodies are seen, which represent arrangement of cells in 2 rows with palisading of the nuclei.1
Subtypes
Neurofibroma
Plexiform neurofibroma
Craniomaxillofacial lesions can be divided into massive plexiform, cranioorbital, and cervical neurofibromas.5
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


