Advances in the methods used to analyze DNA, RNA, and proteins are transforming the practice of medicine and research. Traditional methods to assess individual genes or proteins are complemented by advanced techniques allowing genomic or proteomic analysis of populations of cells or individual cells. The functions of genes and encoded proteins are determined in vivo using animal models that allow control of when and where genes are expressed. Molecular techniques enable gene-based therapies in which genes are corrected, supplemented, or suppressed for therapeutic benefit. The future promise of precision medicine depends on our ability to use and develop these techniques for molecular analysis.
Advances in molecular biology are rapidly changing our understanding of skin biology and disease. Increased knowledge is being translated into new molecular diagnostic tests that are transforming the clinical practice of dermatology. Molecular analyses are currently being employed to diagnose genodermatoses , cutaneous infections , melanomas , lymphomas , and inherited or autoimmune blistering disorders . In order to use these tests in a prudent manner, dermatologists nowadays have an even greater need to understand basic concepts in molecular biology. This knowledge is also leading to the development of new targeted therapies, many of which require determination of the molecular basis for disease in order to appropriately implement treatment . In addition, molecular analysis is being used to screen for drug efficacy and susceptibility to adverse reactions to certain medications .
The goal of this chapter is to outline basic concepts and methodologies of molecular biology and to give practicing dermatologists a fundamental appreciation of what their colleagues are doing in the laboratory and how their own clinical practice is changing as molecular approaches are implemented into patient care.
Experimental Techniques
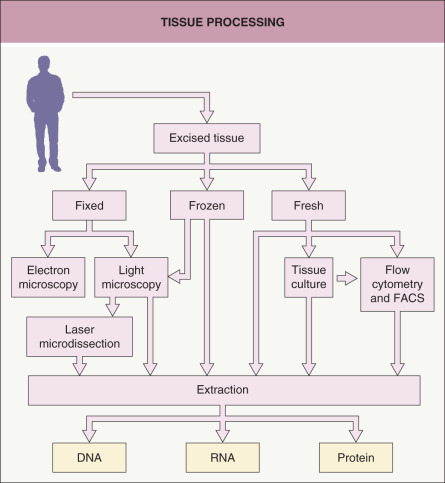
Tissue Processing
Dermatologists are accustomed to placing a biopsy specimen in formalin followed by paraffin embedding and staining tissue sections with hematoxylin and eosin. However, for molecular analysis, this is just one of several possible starting points for tissue processing ( Fig. 3.1 ). Placing the sample in formalin (for light microscopy) or glutaraldehyde (for electron microscopy) before processing and sectioning allows the sample to be analyzed histologically in a variety of ways, including by routine staining, immunohistochemistry, or in situ hybridization. The advantage of this approach is that the cells are preserved in a very stable fashion, with well-preserved architecture, for later analysis. Disadvantages are that the cells are no longer living and the fixation procedure can place limits on the methods that can be used to analyze DNA, RNA, and protein from cells of interest. Nonetheless, PCR analysis of DNA is increasingly being performed on formalin-fixed, paraffin-embedded tissue, especially for infectious diseases and detection of gene rearrangements in lymphoproliferative disorders.
Fig. 3.1
Tissue processing.
A tissue sample can be processed in various ways for the analysis of DNA, RNA, or protein. FACS, fluorescence-activated cell sorting.
In order to preserve molecules in a more native state, the specimen may be snap frozen. Cryosections prepared from such samples have lower quality tissue architecture than do permanent sections from formalin-fixed, paraffin-embedded tissues, but this form of tissue preservation may allow for better analysis of DNA, RNA, and protein. One may also directly process fresh or frozen tissue with buffers and reagents in order to extract DNA, RNA, and protein from the whole tissue. The advantage of performing extractions from whole tissue is that the DNA, RNA, and protein will be fresh and of high quality. The disadvantage is that the extracts will not be from a pure population of cells.
Another approach is to culture cells obtained from fresh tissue. The desired lineage of cells (e.g. keratinocytes, fibroblasts, immunocytes, endothelial cells) can be propagated in vitro by employing selective culture media and isolation techniques. Culturing cells allows one to obtain more cells than in the original sample, and then the cells can be exposed to various conditions. However, the culturing process may change fundamental characteristics of the cells, so that they do not accurately represent the cells in vivo .
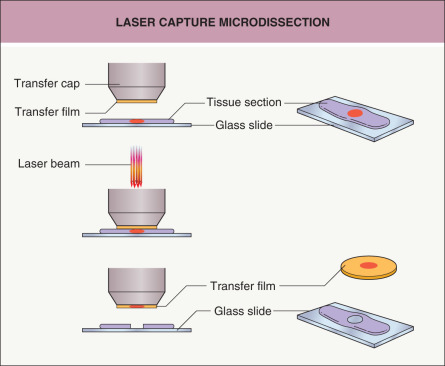
One method for isolating a pure population of cells from a tissue section is to use laser microdissection ( Fig. 3.2 ). A laser is used to retrieve or cut individual cells from a section on a slide while the specimen is viewed with a microscope . An advantage of microdissection is that a very pure population of cells is obtained that has been precisely identified under the microscope. One limitation is that the total number of cells isolated is relatively low, as each cell has to be individually captured by the laser. Consequently, the procedures to extract DNA, RNA and protein from these pure (but few) cells have to be robust.
Fig. 3.2
Laser capture microdissection.
Laser capture microdissection is one method of microdissection used to selectively procure individual cells or clusters of cells from tissue sections. A cap coated with a thermoplastic transfer membrane is placed directly over the tissue section. The operator visually identifies the cells of interest and triggers a low-energy infrared laser to melt the transfer film onto cells of interest. The cap is then lifted from the section to separate the selected cells from the remainder of the tissue section.
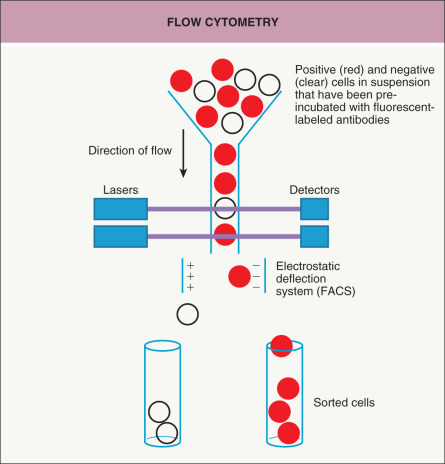
A way to isolate and analyze cells in suspension is to use flow cytometry ( Fig. 3.3 ) . Cells are passed individually in a stream through a set of lasers and electronic detectors capable of measuring multiple parameters for each cell. As an initial step, cells are typically incubated with fluorescent-labeled antibodies that recognize cell-surface markers so that a heterogeneous population of cells can be characterized on a cell-by-cell basis for levels of expression of each marker. Fluorescence-activated cell sorting (FACS), which utilizes an electrostatic deflection system, can also be used to obtain pure populations of cells with specific desired characteristics. Flow cytometry permits the measurement of multiple characteristics of individual cells at a rapid rate. However, with the exception of peripheral blood or bone marrow cells, a significant limitation is that it requires the cells to be in suspension. Recently, the ability to analyze 40 or more parameters for each cell has been accomplished by combining the principles of flow cytometry and elemental mass spectrometry in what is call mass cytometry .
Fig. 3.3
Flow cytometry.
Cells in suspension, pre-incubated with fluorescent-labeled antibodies, flow single file in a liquid stream through lasers. Detectors measure the fluorescence intensities of each cell. In fluorescence-activated cell sorting (FACS), the single cells are divided into charged droplets that are separated into different tubes based on the marker(s) of interest as they pass through an electrostatic deflection system. Flow cytometry is commonly employed in the evaluation of patients with cutaneous B- and T-cell lymphomas.
The Foundations of Molecular Techniques for Analyzing DNA, RNA, and Protein
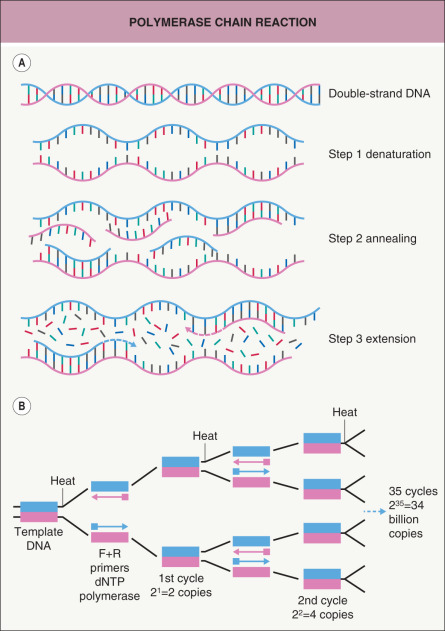
The concepts behind molecular biology are simple and unifying. In general, they consist of extracting the molecules of interest, amplifying them to measurable amounts, and detecting them. Polymerase chain reaction (PCR) is a standard technique for amplifying DNA ( Table 3.1 ; Fig. 3.4 ) . The PCR-amplified DNA, typically 50 to 2000 base pairs in size depending on the primers designed for a particular sequence, can be detected in a gel using an intercalating dye that fluoresces with ultraviolet light. The nucleotide sequence can then be determined via automated fluorescence sequencing techniques ( Table 3.2 ; Fig. 3.5 ). This simple and relatively inexpensive approach is still widely used. However, it is being supplanted by massively parallel sequencing, also known as next-generation sequencing, in which millions of fragments of DNA are sequenced in a single run (see Table 54.6 ) .
Table 3.1
Polymerase chain reaction.
POLYMERASE CHAIN REACTION
Purpose
•
Amplify a specific piece of DNA from a complex mixture
Requirements
•
Need to know sequence of the DNA of interest (at least of its ends)
Underlying concepts
•
Double-stranded DNA can be melted or unwound to single strands with increased temperature ( Fig. 3.4A ); when cooled, the single strands come back together to form double strands (hybridize) if the nucleotide sequences are complementary *
•
During the hybridization process, when two complementary strands bind to each other, the A nucleotides on one strand bind to T nucleotides on the complementary strand, whereas C nucleotides of one strand bind to G nucleotides on the complementary strand, and vice versa
•
In PCR, short DNA strands called oligonucleotide primers are designed for hybridization to specific sequences in the template DNA
Oligonucleotide primers are designed to hybridize to specific sequences at each end of the DNA of interest. These primers are added to a reaction vessel mixture containing the template DNA along with a thermostable DNA polymerase, the nucleotides dATP (A), dTTP (T), dGTP (G) and dCTP (C), and buffer
•
The reaction vessel is placed in a thermal cycler, which controls the temperature of the reaction through many cycles
•
Each cycle contains the following steps: (1) denaturation; (2) primer annealing or primer hybridization; (3) primer extension; and (4) repeat of the complete cycle of PCR 30–40 times
Benefits
•
PCR is simple and rapid
•
Because the PCR product is exponentially increased, it is extremely sensitive in amplifying low amounts of DNA. Each cycle increases the number of PCR products twofold. The total number of PCR products after n cycles will be 2 n
Limitations/errors
•
Because of its high degree of sensitivity, laboratory contamination of a DNA sample by trace amounts of the PCR product can cause misleading results
•
Primers used for PCR can anneal to sequences that are similar, but not identical, to the sequence of interest. This can be countered with “hot start” techniques (DNA polymerase prevented from acting until after first denaturation step) or nested PCR (after the PCR amplification, repeat the PCR amplification using a second set of primers that hybridize to sequences inside first set of primers). In nested PCR, the second set of primers will only hybridize to correct PCR products resulting from the first PCR amplification
•
DNA polymerase occasionally incorporates incorrect nucleotides. For sequencing by PCR, polymerases that possess proofreading enzymatic activity can be used. This also allows the generation of even longer PCR products, up to ~50 kb long
Experimental applications
•
DNA can be amplified either for detection of a specific sequence or for cloning that sequence
•
PCR can be used to label DNA with fluorescent or radioactive nucleotides
•
PCR can be used for rapid haplotype analysis
Modifications/alternatives
•
Quantitative PCR – the amount (number of copies) of a specific piece of DNA can be quantified by a variety of methods utilizing PCR. A relatively simple and high-throughput method, called real-time quantitative PCR, uses a specially designed thermal cycler that measures the amount of PCR product formed after each cycle (see Fig. 3.6B ). The higher the level of DNA, the earlier the product can be detected. This can be used to measure genetic changes in cancer such as gene amplification, to quantify the amount of residual cancer following treatment, or to quantify the amount of a pathogen in a sample. Modifications of this procedure can allow discrimination of gene polymorphisms
•
Southern blot, in situ hybridization, comparative genomic hybridization
* Hybridization actually forms the basis of several techniques in molecular biology, as the two strands can be DNA:DNA (PCR, Southern blotting), DNA:RNA (Northern blotting, in situ hybridization), or RNA:RNA.
Fig. 3.4
Polymerase chain reaction.
A Each cycle contains the following steps: (1) denaturation – separate the two strands of DNA by heating to >90°C; (2) primer annealing or primer hybridization – allow the oligonucleotide primers to bind to the template DNA by cooling to 50–65°C; (3) primer extension – DNA polymerase catalyzes the addition of nucleotides (A, G, C, T) that are complementary to the DNA template, beginning with the primer and extending 3′ at the optimal temperature of 72°C; and (4) repeat the complete cycle 30–40 times. B Each cycle increases the number of PCR products twofold. The total number of PCR products after n cycles will be 2 n times the original amount. F+R, forward and reverse.
Table 3.2
DNA sequencing.
DNA SEQUENCING
Purpose
•
Determine the sequence or order of nucleotides (A, G, C, T) in a stretch of DNA
Requirements
•
The piece of DNA to be sequenced can be either a PCR product or a cloned piece of DNA present in a plasmid, but it should be pure
Underlying concepts
•
Chain termination (Sanger sequencing), or a variation thereof, is a standard method for DNA sequencing
•
In chain termination, the extension of a new strand of DNA is stopped by the addition of an analogue of dATP, dCTP, dGTP or dTTP (ddATP, ddCTP, ddGTP or ddTTP, respectively) to the sequencing mixture. When the DNA polymerase incorporates the analogue nucleotide instead of the correct normal nucleotide, DNA synthesis is terminated because the polymerase is no longer able to link to the next nucleotide
•
Gel electrophoresis is used to separate the different sizes of DNA fragments that result from chain termination synthesis. The DNA fragments are forced to travel through a gel using an electric current; the smaller molecules are less impeded by the gel and travel faster than larger molecules *
An oligonucleotide primer hybridizes to the DNA to be sequenced and DNA polymerase synthesizes a second complementary strand
•
The synthesis of the second strand is interrupted randomly by the incorporation of the fluorescent nucleotide analogues (ddATP, ddGTP, ddCTP, ddTTP), and the DNA fragments containing this final nucleotide analogue can be identified because each of the four ddNTPs is labeled with a different color fluorochrome
•
The different DNA fragments are electrophoresed through a polyacrylamide gel or capillary tubes
•
The different-length DNA strands terminating with different fluorochrome-labeled nucleotide analogues pass a fluorescence detector and indicate the order of the DNA sequence
Benefits
•
The fluorescent chain termination method is able to rapidly sequence large amounts of DNA with automated analysis of results
Limitations/errors
•
Routinely sequence only ~500 bases per run
•
Difficulty with G+C-rich regions
•
DNA must be high-quality
Experimental applications
•
Determine previously unknown sequence
•
Confirm sequence following the cloning of a DNA fragment of interest and other manipulations
Modifications/alternatives
•
Pyrosequencing
•
Next-generation sequencing (massively parallel sequencing; see Table 54.6 )
* Gel electrophoresis is used in many molecular biologic techniques to separate DNA, RNA or protein molecules of differing sizes.
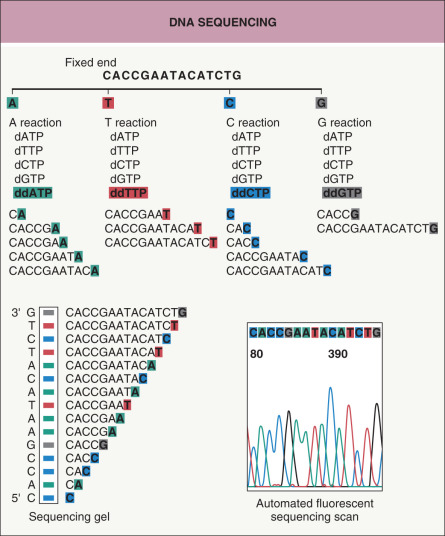
Fig. 3.5
DNA sequencing.
An oligonucleotide primer hybridizes to the DNA to be sequenced and DNA polymerase synthesizes a second complementary strand. The synthesis of the second strand is interrupted randomly by the incorporation of fluorescent nucleotide analogues (ddATP, ddGTP, ddCTP, ddTTP). The DNA fragments containing this final nucleotide analogue can be identified because each of the four ddNTPs is labeled with a different color fluorochrome. Gel electrophoresis is used to separate the different sizes of DNA fragments. The different-length DNA strands terminating with different fluorochrome-labeled nucleotide analogues pass a fluorescence detector and indicate the order of the DNA sequence (see Table 3.2 for more details).
RNA is also easy to purify, but it is much more readily degraded than DNA. Therefore, a typical first step in the analysis of RNA is to convert it into DNA using reverse transcription (RT; Table 3.3 ; Fig. 3.6A ). Following RT, the complementary DNA (cDNA) can be amplified by PCR, as described above. The technique of RT-PCR has also been modified to allow accurate quantitation of very low levels of mRNA . Because the amount of PCR product is monitored throughout each cycle of amplification, this technique is referred to as “real-time” quantitative PCR ( Fig. 3.6B ).
Table 3.3
Reverse transcription PCR (RT-PCR).
REVERSE TRANSCRIPTION PCR
Purpose
•
To amplify mRNA by PCR, the mRNA is first converted to DNA (called complementary DNA or cDNA), followed by PCR amplification of a specific region of the cDNA to detectable levels
Requirements
•
Starting material can be total cellular RNA (including ribosomal, transfer and messenger RNA [mRNA]) or purified mRNA
Underlying concept
•
In order to facilitate studies of RNA, many techniques that study RNA first convert the RNA to cDNA with an enzyme called reverse transcriptase, an RNA-dependent DNA polymerase
Reverse transcriptase can convert mRNA to cDNA by three different methods, depending on the primer used for the initial RT step * . (1) Random hexamer primers contain six nucleotides (6-mer) that have all possible sequence combinations of the dA, dG, dC and dT nucleotides (4 6 possible combinations). These random hexamers will hybridize to the corresponding complementary sequences in the sample RNA. (2) Oligo dT primers contain only dT nucleotides and hybridize to the complementary string of dA nucleotides that are present at the end of mRNA molecules (poly A tail). (3) The third choice is a primer that will only hybridize to a specific mRNA sequence
•
After the mRNA has been converted to cDNA, primers that can hybridize to specific sequences are added and PCR amplification is performed as described in Table 3.1
Benefits
•
As for PCR, RT-PCR is simple and rapid
•
RT-PCR is extremely sensitive in detecting low levels of mRNA transcripts
Limitations/errors
•
If the RNA sample is contaminated with DNA (that contains the gene of interest), a PCR product may be amplified from the DNA even though the corresponding mRNA for the gene is not present. To control for this, RT-PCR of RNA samples can be done with and without reverse transcriptase
•
RNA is fragile and the absence of a specific mRNA transcript may result from RNA degradation during and following extraction. RNA quality can be tested by gel electrophoresis and/or by RT-PCR of housekeeping genes (genes expressed by all cells)
Experimental applications
•
The mRNA gene transcripts can be amplified for subsequent cloning or sequencing
•
The mRNA gene transcripts (instead of the gene) could be analyzed for the presence of mutations
Modifications/alternatives
•
Quantitative RT-PCR – by adding a reverse transcription step to quantitative PCR (see Table 3.1 ), the levels of mRNA transcripts from genes of interest can be quantified.
•
Correspondingly, samples that contain less mRNA transcripts will require more PCR cycles before the exponential phase. Therefore, different RNA samples can be precisely compared by measuring the number of PCR cycles ( x axis) needed to produce a defined amount of PCR product ( y axis) ( Fig. 3.6B )
•
By carefully designing the primers, quantitative RT-PCR can be used to measure the amounts of alternatively spliced forms of a gene
•
Alternative methods to measure RNA levels include digital PCR, in which the sample is partitioned into individual nucleic acids, and Northern blots
* In addition to the RNA, the reaction mixture contains the reverse transcriptase enzyme, an oligonucleotide primer, dNTPs, and buffer.
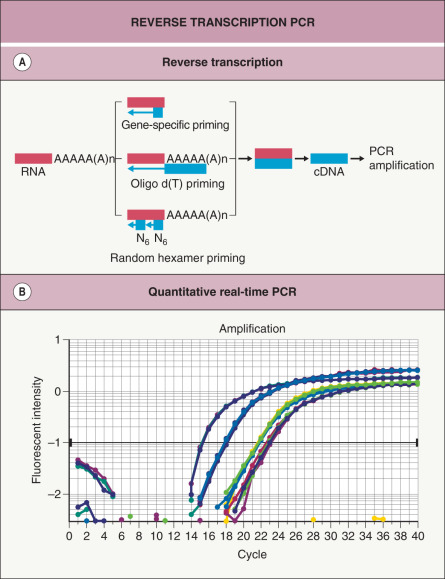
Fig. 3.6
Reverse transcription PCR (RT-PCR).
A Reverse transcriptase can convert mRNA to cDNA in three different ways, depending on the primer used for the initial RT step: (1) random hexamer primers; (2) oligo dT primers; and (3) gene-specific primers (see Table 3.3 for more details). After the mRNA has been converted to cDNA, primers that can hybridize to specific sequences are added and PCR amplification is performed as described in Table 3.1 . B Real-time PCR is able to precisely measure the amount of PCR product continuously ( y axis) after each cycle ( x axis). Each plotted line represents the amount of PCR product present in a different sample. In samples that initially contain more mRNA gene transcripts, real-time PCR will demonstrate an exponential increase in PCR product earlier, after fewer PCR cycles.
The amount of protein is a complex balance of synthesis and degradation controlled at multiple steps, including efficiency of protein translation and post-translational modifications that affect protein stability. One method used to measure levels of protein is referred to as a Western blot ( Table 3.4 ; Fig. 3.7 ); it is also known as an immunoblot because an antibody is employed to detect the protein of interest. In addition to measuring protein levels, Western blot analysis can determine the size of proteins and can reveal whether there are different forms of the protein . Another method commonly used to measure protein levels is an enzyme-linked immunosorbent assay (ELISA; see Table 3.4 ) . An ELISA can provide very exact quantitation of protein levels and may be less expensive and easier to perform than a Western blot.
Table 3.4
Western blot.
WESTERN BLOT
Purpose
•
Western analysis can measure the size and the amount of protein present in a sample
Requirements
•
Western analysis requires an antibody that is specific for the protein of interest (i.e. does not cross-react with other proteins)
Underlying concepts
•
Polyclonal antibodies that react to several epitopes on a protein antigen are obtained by injecting a protein into an animal and later isolating the antibodies from the serum immunoglobulin fraction
•
Monoclonal antibodies that react to only one epitope of a protein antigen are obtained by immunizing mice (or rats, rabbits, or chickens) with the antigen, then fusing the animal’s reactive lymphocytes with an immortal myeloma cell line to create cells that are able to provide antibodies indefinitely
•
A secondary antibody is used to detect the protein or the primary antibodies bound to the protein. These detection antibodies can be visualized by attaching a fluorescent probe or by attaching an enzyme that can produce either light or a color by enzymatic action on a substrate
A solubilized protein mix is separated on a polyacrylamide gel and transferred electrophoretically to a membrane. The membrane is then soaked in a buffer containing the antibody. Bound antibody is detected by a chromogenic or chemiluminescent assay
Benefits
•
Western analysis is a simple and sensitive method to detect and quantify proteins present in a complex mixture
•
Western analysis can determine the molecular weight of a specific protein relative to standard controls
Limitations/errors
•
Proteins are subject to degradation during extraction. The use of protease inhibitors in the extraction buffer helps to prevent degradation
•
To perform a Western analysis, one must possess a highly specific antibody that can recognize denatured proteins
•
Western blots may contain a high background of nonspecific staining. To correct this, blocking agents such as bovine serum albumin or milk protein are used
•
Large proteins transfer poorly from the gel to the membrane, and small proteins may transfer through the membrane without binding. Adequate transfer to the membrane can be accomplished by controlling the duration of the transfer procedure
Experimental applications
•
Detection, quantification, and characterization of a specific protein
•
Identification of antibody activity to a known antigen
Modifications/alternatives
•
Dot blot – a drop of the protein mixture is placed on a paper membrane and the protein of interest detected with antibodies, as in the Western blot. The disadvantage of this technique is that, owing to the elimination of size separation by gel electrophoresis, specific binding to the protein of correct size cannot be distinguished from nonspecific background binding to proteins
•
Immunoprecipitation (IP) – the specific antibody is added to the protein mixture and the resulting antibody–protein complexes are then isolated. Because proteins are not first denatured in immunoprecipitation, they can be detected in a more native configuration
•
IP–Western – protein–protein interactions can be studied by first immunoprecipitating the protein with one antibody, bringing down a protein complex. The protein complex is then separated on a polyacrylamide gel, followed by Western blotting to detect members of the protein complex
•
Enzyme-linked immunosorbent assay (ELISA) – this is a sensitive and specific method for quantifying the amount of a protein. The protein of interest is captured on a plate coated with a monoclonal antibody and other proteins are washed away. The protein is then detected using a second antibody that has been modified for detection using a colorimetric or luminescent assay
•
Immunohistochemistry – this is used to visualize the cellular localization of a protein. An antibody to the protein of interest is applied to a tissue section. The antibody is detected using a secondary antibody coupled to an enzyme that reacts with a substrate to produce a colored precipitate (see Ch. 0 )
Only gold members can continue reading. Log In or Register to continue