Mechanisms of Autoimmune Disease: Introduction
|
Introduction
Autoimmune diseases including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) are relatively common disorders.1 Although the underlying etiologies of these illnesses are still elusive, they arise in the context of a break in the immune tolerance to self.2,3 The mechanisms for abrogation of immune self-tolerance appear to be multifactorial, including genetic and environmental, that work in concert to initiate the eventual hallmarks of disease: unregulated immune activation against self-antigens and subsequent tissue destruction. Immune activation against self-antigens is clinically manifest by the presence of autoantibodies and autoreactive T cells.1,2 Based upon their autoantigenic targets, autoimmune diseases can be classified into organ-specific and systemic autoimmune processes. For example, Grave’s disease, with its autoantibodies against the thyroid-stimulating hormone (TSH) receptor, is a typical example of organ-specific autoimmune disease, as is type I diabetes mellitus (DM) with its autoantibodies and autoreactive T cells directed against components of pancreatic β cells, whereas SLE with its characteristic autoantibodies against ubiquitous nuclear antigens is a good example of a systemic autoimmune disease. Although adaptive immune cells such as B cells and T cells recognize self-antigens and hence dominate the phenotype of the patient with autoimmunity, other immune components including antigen-presenting cells (APC) and complement are involved in various steps from initiation of the autoimmune response to tissue destruction.4–6 In this chapter, the contributions of these components to the development of autoimmune diseases will be discussed, focusing on the latest discoveries.
Genetics
Autoimmune diseases are polygenic, involving both major histocompatibility complex (MHC) and non-MHC genes.7,8 Yet, the concordance rate of autoimmune diseases in monozygotic twins is not 100%, ranging from 10% to 50%, including in RA and SLE,7,9 indicating a significant role for nongenetic factors in the development of these disorders. Nevertheless, genetic influences are strong. For instance, the contribution of alleles of MHC to the development of autoimmunity has been known for more than 30 years. An increased frequency of HLA (human leukocyte antigen)-DR4 has been observed in some patients with RA.8 Using more sophisticated molecular techniques, the sequence of genetic alleles encoding HLA loci have been typed, with the resultant demonstration that the HLA-DRB1 gene is highly polymorphic and such polymorphisms can affect the binding of peptides to HLA molecules and the contacts between the T-cell receptor (TCR) and the HLA molecule.8 Thus, the associations between autoimmune diseases and particular HLA molecules can be explained by a model where disease susceptibility is determined by differences in the ability of different HLA alleles to present autoantigenic peptides to autoreactive T cells.2,8 However, the genetic effect of MHC on disease propensity is broader. As an example, a recent study genotyped a panel of 1,472 single nucleotide polymorphisms (SNPs) across the 3.44 megabase (Mb) classic MHC region in 10,576 DNA samples from patients with autoimmune diseases including SLE, RA, and multiple sclerosis, and from appropriate controls.10 The results of this study showed multiple risk alleles across MHC class I, II, and III in these autoimmune diseases, indicating complex, multilocus effects that span the entire region.
The association of non-MHC genes with autoimmune diseases is now well established,1,7,11 with the recent advent of genome-wide association (GWA) scans that are powerful tools for identifying new genes in autoimmunity.11 These genes include those involved in antigen clearance, apoptosis, cell signaling, and cytokine production as well as in the expression of costimulatory molecules and cytokine receptors.1,2,7,11 For instance, complement components are required for proper clearance of immune complexes, and an increased incidence of homozygous C4 deficiency is found in patients with SLE who have immune complex deposits in damaged organs such as the kidneys.9
Recently, a gene called autoimmune regulator (AIRE) gene has been identified as a candidate gene responsible for the development of autoimmune diseases.12,13 AIRE is a transcription factor that regulates the ectopic expression of proteins, normally expressed in peripheral tissues, in the thymus, allowing for thymic expression of the latter and subsequent negative selection of self-reactive thymocytes before they migrate as mature T cells to the secondary lymphoid organs such as the spleen and lymph nodes. An alteration in thymic expression of AIRE can lead to increased generation of autoreactive T cells due to their impaired negative selection.14 Indeed, patients with mutations of the AIRE gene develop a syndrome called autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) or autoimmune polyglandular syndrome 1 (APS-1) that is characterized by chronic mucocutaneous candidiasis, hypoparathyroidism, and Addison’s disease.15 In addition, patients with this condition often have other organ-specific autoimmune diseases including type 1 DM, autoimmune thyroid diseases, gonadal failure, vitiligo, alopecia, dystrophy of dental enamel and nails, and pernicious anemia.15
An increased incidence of autoimmune diseases has been reported in individuals with a particular variant of genes that affect T-cell activation. These include the cytotoxic T-lymphocyte antigen-4 (CTLA-4) gene and the protein tyrosine phosphatase 22 gene (PTPN22)1,11,16 (see below). CTLA-4 is a key immunoregulatory molecule that restrains T-cell activation. It is expressed on T cells upon TCR stimulation by antigen on APC and competes with CD28, a positive costimulatory molecule also on T cells, for binding to CD80 and CD86 expressed on APC.17 By contrast to CD28 engagement by CD80 and CD86, binding of CTLA-4 to the latter molecules inhibits T-cell activation. The association of SLE and RA with SNPs in the CTLA-4 promoter and coding region has been reported. These polymorphic sites include −1722 position of the CTLA-4 promoter and +49 position of exon-1,18,19 although the functional consequence of such gene polymorphisms is not clear yet. CTLA-4 polymorphisms also have been linked to autoimmune endocrinopathies; for example, a decrease in the alternate splice product of CTLA-4 that is the soluble form of CTLA-4 is found in individuals with CTLA-4 SNP CT60 G/G which is linked with increased susceptibility to Grave’s disease, autoimmune hypothyroidism, and type 1 DM.20 This finding suggests a possible functional role for CTLA-4 polymorphisms in autoimmune diseases.
In a manner analogous to CTLA-4, a protein called programmed cell death 1 (PD-1) is expressed on T cells providing an inhibitory signal upon their activation. This molecule is also expressed on other immune cells including B cells and myeloid cells.1,21 PD-1 is encoded by the PDCD1 gene, with a SNP in its fourth intron associated with autoimmunity, including SLE and RA.22–24 Of interest, this disease-associated SNP impairs the binding of the hematopoietic runt-related transcription factor 1 (RUNX1), leading to aberrant expression of PDCD1 gene and subsequently uncontrolled T-cell activation.22 The association of genetic polymorphisms of CTLA-4 and PD-1, molecules involved in regulating T-cell activation, with autoimmune diseases supports the notion that regulation of T-cell activation by these costimulatory molecules is a critical checkpoint for the development of autoimmunity.
The protein tyrosine phosphatase 22 (PTPN22) gene is another recently identified gene that is associated with autoimmune diseases.1 A variant of the PTPN22 gene, which encodes a tryptophan at codon 620 (620W) instead of an arginine (wild-type), has been found to be associated with an increased risk of RA, SLE, type 1 DM, and Grave’s disease.1 Such a mutation appears to have functional consequences, since the PTPN22 gene encodes lymphoid tyrosine phosphatase (LYP) that modulates the activation of Lck and other kinases involved in TCR signaling.25 Indeed, the PTPN22 620W allele appears to potentially increase the inhibition of TCR activation by downregulating Lck activation.25 Thus, it still needs to be clarified how this allele promotes the development of autoimmunity.
Recently, multiple genes encoding molecules involved in tumor necrosis factor (TNF) receptor signaling have been found to be associated with autoimmunity.11 The best known is TNFAIP3 that encodes TNF inducible protein A20 that is a negative regulator of TNF-induced NF-kB signaling pathways. SNP markers in the gene region near the TNFAIP3 locus have been associated with RA, psoriasis, and SLE.26–28 The gene encoding TNFAIP3-interacting protein 1 (TNIP1) that interacts with TNFAIP3 is associated with psoriasis and SLE,27,29 with an association of the TRAF1 (TNF receptor-associated factor 1) gene with RA also reported.30
Polymorphisms in several cytokine receptors are associated with autoimmunity.11 The IL-23 receptor complex consists of IL-23R and IL-12Rβ1 subunits, with the latter shared with the IL-12 receptor complex that is composed of IL-12Rβ1 and IL-12Rβ2 subunits. The IL23R receptor gene is associated with inflammatory bowel disease and psoriasis.31,32 This is an intriguing point since IL-23 is involved in regulating the development of T helper 17 (Th17) cells that produce the potent proinflammatory cytokine IL-17. In fact, IL-17 is found in psoriatic skin lesions.33 Likewise, the IL12B gene that encodes IL-12β (the p40 subunit of IL-12) has been found to associated with psoriasis.34
In addition to genes expressed by T cells, those involved in humoral immune responses also appear to be associated with autoimmunity. The receptors for immunoglobulin G (IgG) Fc portion (FcγR) are expressed on B cells, macrophages, monocytes, granulocytes, and dendritic cells (DCs). There are eight genes identified for the human FcγR: FCGRIA, FCGRIB, FCGRIC, FCGRIIA, FCGRIIB, FCGRIIC, FCGRIIIA, and FCGRIIIB.35 The binding of FcγRs to IgG, except for that to FcγRIIb, results in a broad spectrum of activating cellular responses including phagocytosis, cytolysis, cytokine production, and degranulation that lead to inflammation. By contrast, FcγRIIb physiologically serves as an inhibitory receptor that downregulates the effector function of cells.35 An increased frequency of some variants of FCGR genes has been reported in humans with autoimmune diseases including SLE and RA.35–39 FcγRIIa with an arginine at position 131 has greater signaling in response to IgG1 engagement than FcγRIIa with a histidine at the same position, suggesting that FcγRIIa gene polymorphisms have physiologic relevance.39 Of interest, FcγRIIa with an arginine has been reported as a susceptibility factor for SLE in some ethnic groups.35,40 Similarly, a variant of FcγRIIIa with a phenylalanine at position 158 may be a susceptibility factor for SLE and RA.36,38 In addition, genetic polymorphisms of the inhibitory FcγRIIb have been reported in patients with SLE.37,41,42 The best known such polymorphism is a substitution of an isoleucine at position 232 (187 by counting from the N-terminus of the mature protein) with a threonine.37,41,42 The latter genotype is more commonly reported in patients with SLE. While the biologic implication of such an alteration on the development of SLE is as yet unclear, functioning inhibitory FcR are necessary for dampening autoimmunity in murine lupus.43
Overall, genetic studies on MHC genes and non-MHC genes support the notion that some patients with autoimmune diseases have an increased genetic risk for the development of autoimmune diseases secondary to genetic alterations affecting immune cell function. However, such alterations are not always observed in patients with autoimmune diseases and ethnic differences in genetic susceptibility are not uncommon. These findings support the idea that autoimmune diseases are polygenic and that environment factors are also involved in their development (Fig. 154-1).
Figure 154-1
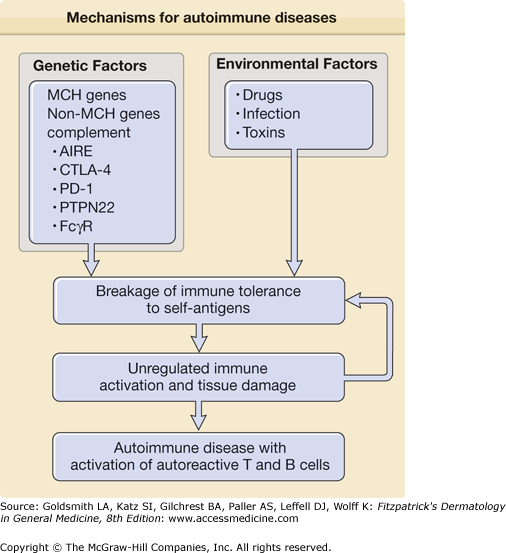
Mechanisms for autoimmune diseases. The figure shows potential mechanisms for initiation of autoimmune diseases including genetic and environmental factors that work in concert to initiate breakage of immune tolerance to self-antigens. Subsequently, unregulated immune activation and tissue damage arise, leading to the development of autoimmune disease with activation of autoreactive T cells and B cells with autoantibody production. Abbreviations: AIRE (autoimmune regulator), CTLA-4 (cytotoxic T-lymphocyte antigen-4), PD-1 (programmed cell death 1), PTPN22 (protein tyrosine phosphatase 22), and FcgR (receptor for immunoglobulin G (IgG) Fc portion). See details in text.
Tolerance and Autoimmunity
Intact mechanisms of self-tolerance are necessary to prevent the development of autoimmune diseases.2 Self-tolerance is generated and maintained through the capacity of the immune system to distinguish self-reactive B and T cells from nonself-reactive cells. These processes begin during the development of lymphocytes in the bone marrow and thymus, respectively.3 Such selective processes are controlled by the binding affinity of antigen receptors to self-antigens during lymphocyte development. B and T cells expressing antigen receptors with affinity for self-antigens are altered, either becoming nonreactive to antigenic stimuli (through a process called anergy induction) or changing their antigen receptors so that they no longer bind self-antigens (a process called receptor editing, operative in self-reactive B cells), or if they bind too strongly to self, they are simply killed via apoptosis. These interventions lead to central tolerance to self-antigens.3 A defect in central tolerance can cause autoimmune diseases as evidenced by the development of APS-1 in humans with an alteration in the AIRE gene (see Section “Genetics”) that is involved in negatively selecting autoreactive T cells.44,45
Despite removing or modifying autoreactive lymphocytes in the thymus and bone marrow by selective processes, some lymphocytes reactive to self-antigens still mature and enter the peripheral immune system; i.e., negative selection is not perfect, probably because if it were, potentially beneficial T and B cells would never make it past central selection in the thymus and bone marrow, respectively. The autoreactive T and B cells that escape central selection can potentially be activated upon recognition of self-antigens; thus, peripheral tolerance mechanisms need to be available to avoid the development of autoimmunity. Such mechanisms are several, and involve minimizing contacts of autoreactive cells with self-antigens (ignorance of the self-reactive lymphocyte), not providing appropriate signals for their activation, terminating activated autoreactive cells by regulatory molecules such as PD-1 or CTLA4, and/or actively suppressing autoimmune responses by other T cells.2 In critical organs like the brain, eyes, and gonads, immunological ignorance is a principal mechanism for avoidance of autoreactivity via the provision of anatomical barriers separating the tissue and lymphocytes. Activation of the latter can be further prevented by the relative paucity of APCs in these organs and the minimal expression of MHC molecules in these tissues.46,47 These sites are termed immunologically privileged since even tissue grafts do not elicit immune responses therein. Immunologically privileged sites have additional mechanisms that prevent immune activation, including production of TGF-β, which can suppress immune responses such as in the anterior chamber of the eyes, and constitutive expression of Fas ligand that induces apoptosis of infiltrating activated lymphocytes.46,47
Nevertheless, since autoantigens are expressed on some tissues and autoreactive T (and B) lymphocytes are present in the circulation, there is still a reasonable chance for the latter to recognize autoantigens expressed by MHC molecules on APC. However, such an autoantigenic signal through potentially self-reactive antigen receptors is not enough to activate lymphocytes, as additional signals (second signals) provided by APCs, such as CD80 and CD86, to CD28 on T cells are required for proper activation.17,48 When proper costimulation is not provided by antigen presenting cells, autoreactive T cells that are only signaled through the TCR undergo apoptosis or become anergized. As CD80 and CD86 upregulation on APCs requires an inflammatory stimulus provided to the APC such as that provided by a pathogen during infection, presentation of self-antigens in the absence of inflammation does not lead to autoreactive T cell activation. By contrast, peptides of pathogens lead to cellular activation as they are presented to the immune system in the appropriate inflammatory context. Of course, this regulatory control on autoimmunity may be bypassed if in a similar manner self-antigens are presented at a site of inflammation. Fortunately, this appears to be a relatively unusual event, and autoimmunity invoked by this mechanism is typically avoided.
Autoreactive lymphocytes that manage to get activated in the periphery can be also deleted by apoptosis (programmed cell death) and/or their activation terminated by inhibitory molecules expressed on autoreactive lymphocytes. Fas ligand, or Fas, expressed on T cells is largely responsible for the former.49,50 The latter process can be achieved through CTLA-4 and PD-1 expressed on activated T cells that inhibit and downregulate T cell activation.17,21 The potential roles for CTLA-4 and PD-1 in controlling autoimmunity have been demonstrated by mouse studies where these genes have been genetically eliminated, for example, with resultant autoimmunity.51,52 In addition, genetic polymorphisms in these molecules have been reported to be associated with autoimmune diseases including SLE and RA (see Section “Genetics”). More importantly, there have been attempts to treat autoimmune diseases by modulating CTLA-4 and PD-1 inhibitory pathways.53,54 Indeed, abatacept, a recombinant fusion protein comprising the extracellular domain of human CTLA4 and a fragment of the Fc domain of human IgG1, is available for the treatment of RA.53 This drug, like CTLA4, competes with CD28 for CD80 and CD86 binding; however, in contrast to CTLA4 engagement that leads to downregulation of T cell activation, it blocks T cell activation and thus selectively modulates T cell activation.
The activation of autoreactive lymphocytes is also actively suppressed through other mechanisms. The most appealing such mechanism is immune suppression through subsets of T cells called regulatory T cells (Tregs), discussed below.
The concept that certain T cells can regulate immune responses dates back to early 1970s. These cells, that Gershon initially called “suppressor cells,”55 remained largely undefined and their mechanism of suppression not identified. After their original discovery, they remained out of vogue until over a decade later when others including Sakaguchi reported modulation of immune responses by CD4+ T cells expressing CD25 (IL-2 receptor alpha chain)56 as well as by T cells producing TGF-β and IL-10.57–59 Cells with immune regulatory functions are now called regulatory T cells (Tregs). Several subsets of Tregs have been identified thus far, including: (1) naturally occurring CD4+ CD25+ Tregs expressing the transcription factor protein forkhead box P3 (Foxp3); (2) CD4+ T cells producing IL-10 called type 1 regulatory T cells (Tr1); (3) CD4+ T cells producing TGF-β named T helper 3 cells (Th3); and (4) CD8+ T cells producing IL-10 or TGF-β.60,61
Among the best characterized Tregs are naturally occurring CD4+ CD25+ Foxp3+ cells (FOXP3+ Treg) that are produced in the thymus. The potential role for these Tregs in controlling the development of autoimmunity was initially demonstrated by studies where mouse T cell suspensions were transferred into congenic athymic nude mice. If the latter animals received such suspensions depleted of CD4+ CD25+ T cells, they developed an autoimmune syndrome including thyroiditis, gastritis, insulitis, sialoadenitis, adrenalitis, oophoritis, glomerulonephritis, and polyarthritis.56 By contrast, athymic nude mice that received whole T-cell suspensions did not develop autoimmunity, indicating a role for CD4+ CD25+ T cells in preventing autoimmune diseases.56 Subsequently, the inhibitory effects of CD4+ CD25+ Tregs in different types of animal models of autoimmune diseases including collagen-induced arthritis (CIA) and autoimmune diabetes were documented.62,63 Yet, not all CD4+ CD25+ T cells have immunoregulatory functions as CD25 is upregulated upon T-cell activation. Thus, the search for other markers that specifically identify these cells has been extensive, and has led to the identification of CD39, CD103, CTLA-4, glucocorticoid-induced TNF receptor (GITR), lymphocyte activation gene 3 (LAG-3), interleukin 7 receptor, and Foxp364,65 as potential markers. The most promising of this group is Foxp3. Disruption of its gene in mice results in autoimmune diseases,66,67 whereas overexpression of Foxp3 in T cells induces expansion of CD4+ T cells with immunosuppressive function.66–68 Furthermore, CD4+ CD25− T cells transfected with FoxP3 acquire immunosuppressive function.66,67 Although it was originally thought that FoxP+ Tregs develop only in the thymus, studies indicate that CD25− CD4+ T cells in the periphery can become FoxP3+ Treg-like cells with immune regulatory function in the presence of T-cell receptor triggering, IL-2, and TGF-β.69 The latter cells are now called adaptive or inducible FoxP3+ Treg (FOXP3+ iTreg) whereas the former cells are called naturally occurring FoxP3 Treg (nTreg).69
Although the exact mechanism(s) by which naturally occurring FoxP3+ Tregs suppress autoreactive T cells is not fully understood, cell-to-cell contact is necessary and CTLA-4 appears to be required.70,71 CTLA-4 expressed on Tregs interacts with CD80 and CD86 expressed on APC as well as on the target T cells72,73; while CD80 and CD86, ligands for CTLA-4, are conventionally expressed on APC as noted above, they have also been found on activated CD4+ T cells.72,73 These molecules likely interact with CTLA-4 expressed on FoxP3+ Treg, providing “outside-in” signaling leading to immune suppression. IL-2 appears to be involved in inducing and maintaining these cells.71 IL-2 can upregulate FoxP3 expression through activating STAT5.74,75 Of interest, CD25, the IL-2 receptor α chain, could be involved as a suppressive mechanism of FoxP3+ Tregs by absorbing IL-2 and subsequently reducing the availability of this cytokine to other T cells.76 These Tregs also express CD39 (ectonucleoside triphosphate diphosphohydrolase 1) and CD73 (ecto-5-nucleotidase). These molecules generate pericellular adenosine with immunosuppressive activity by catalyzing extracellular nucleotides.77 FoxP3+ Tregs can suppress T cell function by inducing the production of the enzyme indoleamine 2,3-dioxygenase (IDO) from DCs. IDO catabolizes conversion of the essential amino acid tryptophan to kynurenine that is harmful to T cells.71,78 Overall, FoxP3 Tregs likely employ multiple mechanisms in suppressing other T cell function.
By contrast to animal studies, the data on CD4+ CD25+ Tregs in human autoimmune diseases is relatively minimal. In patients with type 1 DM, a typical example of an organ-specific autoimmune disease, the frequency of CD4+ CD25+ Tregs has been shown to be significantly lower than in healthy controls and patients with type 2 DM, suggesting a role for these cells in disease development.79 However, in separate studies, no difference in the frequency of CD4+ CD25+ Tregs and FOXP3 gene allelic variation has been reported in patients with type 1 DM compared to healthy controls,80,81 although the inhibitory function of such cells was impaired.81 Similarly, there was no difference in the frequency of CD4+ CD25+ T cells and their functional markers including Foxp3 among patients with APS-II characterized by Addison’s disease, type I diabetes and autoimmune thyroid disease as well as control patients with single autoimmune endocrinopathies and normal healthy donors.82 However, CD4+ CD25+ Tregs from APS-II patients were defective in their suppressive capacity.82 These findings suggest that patients with autoimmune endocrinopathies including type 1 DM and APS-II may have an alteration in the suppressive function of CD4+ CD25+ Tregs.
A role for CD4+ CD25+ Tregs in the development of SLE and RA, typical systemic autoimmune diseases, has also been explored. Most studies on CD4+ CD25+ Tregs in human lupus reported a decreased frequency of this cell subset,83 suggesting their potential role in the development of SLE. In RA, CD4+ CD25+ Tregs from both peripheral blood and synovial fluid have been studied.84–86 The frequency of CD25+ CD4+ Tregs in the latter fluid was higher than that in peripheral blood,85,86 although there was no significant difference in the numbers of CD4+ CD25+ Tregs in peripheral blood between patients and controls.85,86 Of interest, compromised function of CD4+ CD25+ Treg cells in suppressing inflammatory cytokines from CD4+ CD25− T cells was reported in RA.87 Furthermore, such impairment was reversed after treating patients with anti-TNF-α therapy. These findings suggest that patients with RA have a functional, but not a numerical, defect in CD4+ CD25+ Tregs.
In contrast to CD4+ CD25+ Tregs that naturally occur, other regulatory T cells can be induced in vivo and in vitro.61 These cells include Tr1, Th3 (CD4+ T cells producing large amounts of TGF-β) and CD8+
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


