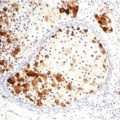
Fig. 44.1
Malignant rhabdoid tumor. Rhabdoid, large round cells with abundant pale eosinophilic cytoplasm containing hyaline filamentous inclusions, eccentric nuclei, and large prominent nucleoli
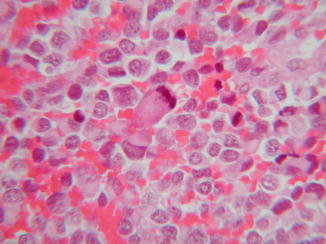
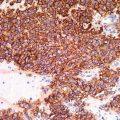
Fig. 44.2
Malignant rhabdoid tumor. Mitotic activity is prominent
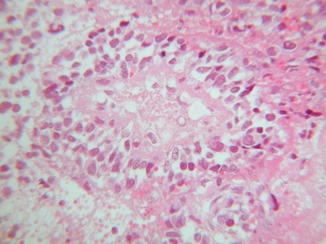
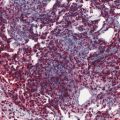
Fig. 44.3
Malignant rhabdoid tumor. Areas of necrosis are present
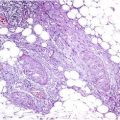
Rhabdoid tumors are often positive for vimentin (>90 %) (Fig. 44.4a), epithelial membrane antigen (EMA) (80 %), and AE1/AE37 (80 %) and can show focal expression of glial fibrillary acidic protein (GFAP), desmin, S-100, neuron-specific enolase (NSE), and actin (Fig. 44.4b). Moreover, 90 % of tumors lack the normal nuclear immunohistochemical expression of the protein INI1 (SMARCB1 or SWI-SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1) derived from the inactivation of hSNF5/INI-1 tumor suppressor gene (22q11.2). On electron microscopy, the tumor cells show a cytoplasmic paranuclear whorl of intermediate filaments containing an entrapped rough endoplasmic reticulum, mitochondria, and lipids.
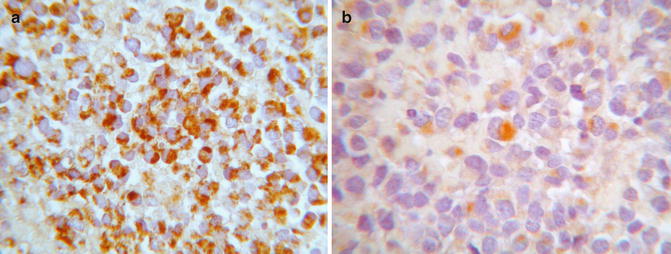
Fig. 44.4
Malignant rhabdoid tumor. (a) Rhabdoid cells are positive for vimentin and (b) focally for actin
Differential Diagnosis
The lack of expression of the leukocyte common antigen (LCA), Wilms tumor 1 (WT1), and NSE allow to rule lymphoma, Wilms tumor, and neuroblastoma. Rhabdoid morphology has been described in squamous cell carcinomas, but lack of expression of both high- and low-molecular-weight cytokeratins excludes an epithelial neoplasm. Moreover, rhabdoid phenotype has been demonstrated in conjunction with epithelioid malignant peripheral nerve sheath tumors, rhabdomyosarcomas, and melanomas. Rhabdomyosarcoma can be diagnosed if markers for striated muscles (MSA, myogenin, MyoD1) are present.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


