Malignant Fibrous, Fibrohistiocytic, and Histiocytic Tumors of the Dermis: Introduction
|
Malignant fibrohistiocytic tumors are a heterogeneous group of mesenchymal neoplasms. These neoplasms occur in the dermis and subcutaneous tissue and may cause diagnostic difficulties. This is at least in part due to the fact that these tumors are relatively rare while at the same time there is a perplexing variety of morphological types and subtypes; thus, the individual physician usually has limited experience in these tumors. By light-microscopic examination cells constituting fibrohistiocytic tumors are characterized by morphologic similarities to fibroblasts and histiocytes. Notably, the term “fibrohistiocytic” only denotes this morphologic appearance, it does not necessarily account for the histogenesis of the respective neoplasm.
The initial descriptions of these tumors were based entirely on hematoxylin and eosin (H&E) morphology and the term “fibrohistiocytic” was chosen in an attempt to provide an organizing principle and nomenclature for this group of soft tissue tumors, composed of cells resembling fibroblasts and histiocytes.
Over the last decades all scientific attempts to demonstrate true histiocytic differentiation in these tumors failed. By means of electron microscopy, immunohistochemistry (IHC), and cytogenetics, it became increasingly obvious that the term “fibrohistiocytic” is a misnomer and falsely unite a heterogeneous group of tumors, many of them are probably unrelated. This notion has been also recently endorsed by the World Health Organization by the use of the terminology “So-called Fibrohistiocytic Tumors” indicating that this term is only used descriptively.1
The symptoms that call attention to a fibrohistiocytic tumor are usually those caused by its presence and growth at its site of origin. In most cases the patient may present with an asymptomatic mass. Thus, a biopsy (either open or large-gauge core needle) is needed to obtain adequate tissue for diagnosis. Care should be taken to ensure that the biopsy does not interfere with subsequent optimal definitive surgery. Moreover, it should be kept in mind that fibrohistiocytic tumors can be very heterogeneous: the smaller the biopsy sample, the more likely it is that only a temporary, working diagnosis becomes possible. This is of course particularly true for a purely cytological examination, since this can only evaluate individual cells. Nevertheless, in the hands of an experienced investigator, the cytological findings in most cases provide enough information to plan the subsequent therapeutic strategy.
The diagnosis of fibrohistiocytic tumors is done principally on the basis of the H&E-stained section. Despite the enormous increase in additional methods available, the H&E section remains the gold standard of morphological diagnosis. When necessary, IHC can be employed; this can reveal cell-line differentiation or antigen expression patterns typical for a particular tumor entity. Likewise, methods of genetic analysis allow the detection of chromosomal translocations, as well as gene deletions or amplifications. These methods are usually exercised when no tumor classification is possible on the basis of the H&E section even in combination with IHC. Chromosomal translocations can be detected in formalin-fixed paraffin-embedded (FFPE) tissue by fluorescence in situ hybridization (FISH) or by reverse transcription polymerase chain reaction (RT-PCR) for the product generated by gene fusion (hybrid RNA). The latter has the advantage of demonstrating not only the translocation, but also the sites of the breakpoints in the genes. Unfortunately, chromosomal translocations are not always diagnostically unambiguous.
Malignant fibrohistiocytic tumors of the dermis represent the malignant end of a spectrum that begins with fibroplasias and benign fibrous tumors. Fibrous lesions are often composed predominantly of stroma, and only in certain high-grade sarcomas the neoplastic nature of the lesion is immediately obvious. However, it should be noted that the identification of defined chromosomal abnormalities in some fibrohistiocytic lesions traditionally regarded as reactive suggests their neoplastic status. Thus, the classification of fibrohistiocytic tumors, like that for other tumor entities, is not static. New aspects or interpretations have to be taken into account and incorporated into clinical practice, i.e., it becomes necessary to update the tumor classification, modify it, or even alter major parts of it.1
A prominent example for such an evolution is malignant fibrous histiocytoma (MFH). MFH as a tumor entity has already been a subject of debate for sometime. Tumors were originally classified as MFH based on the assumption that the histiocytic tumor cells possess the ability to modulate into facultative fibroblasts. Indeed, MFH was regarded as the most common soft-tissue tumor of advanced adulthood, with pleomorphic MFH being the prevalent subtype. Subsequently, however, it was demonstrated that the phenotype of pleomorphic MFH can be adopted by various soft tissue malignancies by loss of differentiation; thus, in the majority of cases there is no independent tumor entity and the cells of pleomorphic MFH are basically dedifferentiated or undifferentiated cells of different origin. By means of more advanced techniques it was possible to demonstrate markers of residual differentiation for the majority of these tumors and thereby to remove such defined soft-tissue tumors from the group of pleomorphic MFH. According to the recommendations of the WHO, the remaining tumors that cannot be further subtyped, i.e., pleomorphic MFH in the narrower sense, should be described as undifferentiated pleomorphic sarcomas (UPS); notably, these very rare tumors are almost never cutaneous lesions. The tumor cells of the second prominent MFH subtype—the myxoid variant—are now understood as fibroblastic cells; thus, the preferred designation today is myxofibrosarcoma (MFS).
With regard to the treatment of “so-called malignant fibrohistiocytic tumors” surgery is the primary treatment of choice. In cases where surgery is not feasible, i.e., large primary tumors or inoperable metastatic disease, beneath chemotherapy and radiation the search for suitable molecularly targeted agents is strongly pursued.2 To achieve this goal, however, a pathogenetically essential molecular target is needed. This strategy up to now was successful in one tumor entity only, dermatofibrosarcoma protuberans (DFSP), which can be effectively treated with tyrosine kinase inhibitors (TKI) targeting the platelet-derived growth factor receptor (PDGFR), for example, imatinib. For all other entities of malignant fibrohistiocytic tumors, clinical studies investigating different targeted agents in an unselective manner did not show any promising results. Here, the key molecular pathways for pathogenesis and maintenance of the different tumor entities first have to be elucidated in more detail to subsequently serve as targets for a molecularly driven therapy.
Dermatofibrosarcoma Protuberans
The incidence of DFSP ranges between 0.5 and 1:100,000, thus it is the most common cutaneous sarcoma. Although DFSP can present at birth or in children, most patients are middle-aged adults. There does not seem to be a gender or racial predilection for the tumor, however, the pigmented variant (Bednar’s tumor) is more common in black patients.
The histogenesis of DFSP is uncertain: fibrohistiocytic, purely fibroblastic, and neural-related differentiation have all been hypothesized. At the molecular level, DFSP is characterized cytogenetically by a reciprocal translocation t(17;22) (q22;q13), or more frequently, a supernumerary ring chromosome composed of hybrid material derived from t(17;22).2 The translocated chromosome and the supernumerary rings contain the same molecular genetic rearrangements. The t(17;22) translocation fuses the gene PDGFB on chromosome 22 with the strongly expressed collagen 1 alpha 1 (COL1A1) gene on chromosome 17. The PDFGB gene encodes the β chain of the PDGF, a ligand for the cell-surface receptor tyrosine kinase PDGFR (both PDGFRβ and PDFRα). PDGFβ is a growth factor that acts as a potent mitogen for a variety of connective tissue cells. The formation of the COL1A1-PFGFB fusion gene results in constitutive production of COL1A1-PDGFβ protein. After translation the COL1A1-PDGFβ fusion undergoes posttranslational processing to form functional PDGFβ. Thus, the production of PDGFβ can lead to autocrine stimulation of the tumor’s own PDGFR, promoting tumor growth and survival. These observations suggest that deregulated expression of PDGFβ, with concomitant autocrine stimulation of PDGFR, is the critical molecular event in the pathogenesis of DFSP. Notably, both fibrosarcomatous transformation of DFSP and some cases of superficial fibrosarcoma (which actually may represent fibrosarcomatous transformed DFSP) have been shown to harbor these fusion transcripts, suggesting a close affinity between DFSP and fibrosarcoma.
In some patients, there is a history of previous trauma; in this regard it should be noted that DFSP has also been described as arising in scars, including surgical, burn, and vaccination sites.
DFSP is a slow-growing lesion that often presents on the trunk and proximal extremities, less frequently on the head and neck of patients. Due to its indolent onset, the patient may present rather late when the tumor is already several centimeters in size. The tumor is often misdiagnosed as a simple scar, keloid, or cyst (Box 125-1). Even when allowed to grow for many years, the tumor usually remains asymptomatic.
Dermatofibrosarcoma Protuberans
|
Atypical Fibroxanthoma
|
Epithelioid Sarcoma
|
The clinical presentation of DFSP is variable. The most common presentation is a firm, indurated plaque, often skin-colored with red–brown exophytic nodules (Fig. 125-1A). Purely plaque-like DFSP never developing nodules or tumors consisting of only a single nodule are rare. Initially the tumor is freely moveable; subsequently, however, larger adherent tumors evolve. At this stage the overlying epidermis may be thinned and telangiectases appear. Bleeding and ulceration are uncommon. DFSP may less frequently present as a nonprotuberant, atrophic (Fig. 125-1B), violaceous lesion resembling morphea or a sclerosing basal cell carcinoma; this form is more common in childhood. The pigmented variant of DFSP is termed Bednar tumor.
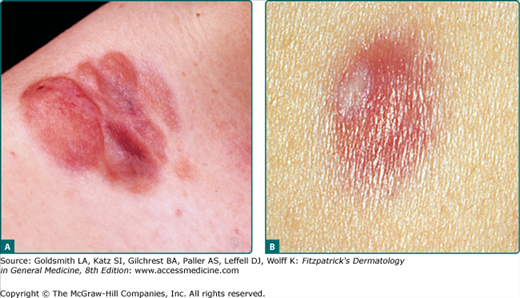
Giant cell fibroblastoma is considered a variant of DSFP, i.e., the juvenile variant of DFSP. Giant cell fibroblastoma typically presents as a dermal or subcutaneous mass, which most frequently involves the trunk, thigh, or inguinal region. It presents much earlier in life than the typical DFSP, often within the first decade, although occurrences in adults have been reported.
DFSP is a dermal proliferation of monomorphous, slender, or slightly plump spindle cells with little pleomorphism arranged in a storiform pattern (Figs. 125-2A and 125-2B). The epidermis is usually uninvolved. The proliferation commonly infiltrates the subcutaneous fat along the fibrous septa, isolating adipocytes to form lucencies (“honeycomb” pattern). The periphery of the tumor is poorly defined, rendering histological control of the surgical margins difficult. In contrast to dermatofibroma, the tumor is much more cellular and usually does not have mature collagen interspersed between fascicles of spindle cells. Mitoses are present; a high mitotic rate may correlate with an impaired prognosis.
Figure 125-2
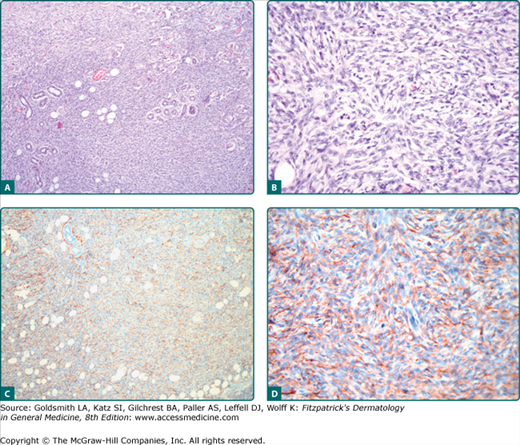
A and B. Dermatofibrosarcoma protuberans showing fibroblastic spindle cells that are arranged in interlacing fascicles. These intersect and bend at acute angles, producing a starburst (storiform) pattern. C and D. Strong expression of CD34 of the tumor cells. Magnification: A and C, 10×; B and D, 20×.
Variable histologic patterns described include myxoid, neuroid, fibrosarcomatous, and granular cell types. The tumor may be relatively monomorphous or may show combinations of various patterns within the original tumor or in the recurrences. The Bednar tumor is rich in melanocytes. Giant cell fibroblastoma is characterized by giant cells, irregular vascular-like space partially ligned by giant cells as well as myxoid to collagenous areas with elongated to stellated cells. Some lesions show overlap between giant cell fibroblastoma and DFSP. The fibrosarcomatous dedifferentiation appears histologically as cellular tumor zones with a fascicular growth pattern, cellular atypia, and numerous mitoses. These atypical cells are arranged in long fascicles in a herringbone (fibrosarcoma-like) pattern; this transformation appears to be related to mutations in the p53 pathway.
A very sensitive, although nonspecific, marker for DFSP is CD34.3 DFSP cells label diffusely and strongly with antibodies to CD34 (Figs. 125-2C and 125-2D) and vimentin; however, CD34 positivity may be lost in nodular regions; particularly the fibrosarcomatous dedifferentiated cellular zones can be negative for CD34. The low-affinity nerve growth factor receptor p75 has been reported positive in DFSP cells. Scattered Factor XIIIa positive cells may be present. Tenascin is negative at the dermoepidermal zone in DFSP. DFSP cells are negative for S100 protein, smooth muscle actin (SMA), desmin, keratins, and epithelial membrane antigen (EMA). In contrast to dermatofibroma, stromelysin 3 is not expressed in DFSP.
DFSP is a low-grade malignant tumor, often infiltrating diffusely through the dermis and into the subcutaneous fat but seldom metastasizing. Increased age, high mitotic index, and increased cellularity are predictors of poor clinical outcome. Approximately 30%–50% of DFSPs recur locally after simple excision. Local recurrences even after wide excision with 1–3-cm margins to the fascia or periosteum are common. Best results can be achieved by means of micrographic controlled surgery (Mohs micrographic surgery) for tumor extirpation. However, it should be kept in mind that by use of frozen sections the histomorphology is not as good as by use of formalin-fixed paraffin embedded sections—in some cases impede correct diagnosis. Metastases of DFSP are extremely rare, usually occurring in the setting of multiply recurrent tumors and tumor progression to fibrosarcoma. Metastases to the lung are most common, with nodal disease the next most common site of spread. Metastatic disease portends a poor prognosis. It has been suggested that tumors with fibrosarcomatous change may have a higher risk of recurrence or metastases. It is estimated that there is a 5% transformation rate from DFSP to conventional fibrosarcoma. The true impact of fibrosarcomatous changes seen within a DFSP remains uncertain.
Standard management of localized disease is complete local surgical resection. With standard excision, margins of 1–3 cm may be necessary to achieve clear margins. However, local recurrence rates are high (13%–52%), especially in fibrosarcomatous DFSP. Risk of local recurrence decreases with increasing surgical margins. Pathologic examination of margins during surgery is helpful in delineating the extent of the tumor. Multiple case series have shown Mohs micrographic surgery to be an extremely effective method of resection of DFSP, with an extremely low rate of local recurrence. As a result, Mohs micrographic surgery represents the preferred surgical approach of many practitioners.
The role of radiotherapy remains unclear. Radiotherapy in the adjuvant setting following surgery is a treatment option particularly when margins are positive or close after maximal resection, if there is concern about the adequacy of negative margins, or if the achievement of wide margins would result in a functional or cosmetic defect.
Targeting the PDGF receptor signaling by receptor TKI, such imatinib or nilotinib, has demonstrated remarkable clinical success in the setting of advanced, unresectable primary tumors or metastatic disease.4
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


