Introduction
Lymphedema is a chronic, debilitating disease resulting from infections, obesity, congenital abnormalities, or traumatic injuries. Congenital abnormalities of the lymphatic system resulting in lymphedema are referred to as primary lymphedema. In contrast, lymphedema resulting from trauma, injury, infections, or other external factors is known as secondary lymphedema. Regardless of the type, patients who have lymphedema experience swelling, pain, recurrent infections, and have impaired quality of life.
In the Western world, the most common cause of lymphedema is cancer treatment and iatrogenic lymphatic injury (i.e., secondary lymphedema). Breast cancer, due to its high incidence, is the most common cause of secondary lymphedema afflicting as many as 50% of patients who undergo axillary lymph node dissection. The advent of sentinel lymph node biopsy has decreased the need for lymph node dissection in many patients, and as a result, has decreased the risk of lymphedema. However, it is important to note that lymphedema can still develop in 5%–7% of patients who undergo sentinel lymph node biopsy. In addition, lymphedema is not a disease that is restricted to breast cancer survivors, developing in nearly 1 in 6 patients who undergo surgical management of gynecological/urological tumors, sarcoma, or melanomas. As a result, lymphedema is prevalent, affecting nearly 6 million Americans. However, this number pales in comparison to worldwide figures, with an estimated 200 million individuals who suffer from lymphedema as a result of chronic parasitic infections. Because the mainstay of current treatment is palliative measures such as compression garments and physical therapy, it is incumbent on the medical community to develop effective medical and surgical treatment options for this disease. This chapter will review the anatomy and physiology of the lymphatic system and outline current advances in the surgical management of lymphedema.
Anatomy of the Lymphatic System
Lymphatic Capillaries and Collectors
The lymphatic system has several functions, including absorption of interstitial fluid and macromolecules, migration of immune cells and regulation of inflammation, initiation of adaptive and innate immune responses, intestinal fat absorption, and cholesterol metabolism among others. Interstitial proteins, immune cells, and particulate matter that are too large to enter the capillary venous system are passively and, in some cases, actively absorbed by initial lymphatics. The initial lymphatics drain into successively larger collecting lymphatics that then actively transport these substances via coordinated pumping of smooth muscle cells that cover the collectors and one-way valves that prevent backflow of lymph ( Fig. 44.1 ). Lymphatic fluid is eventually returned to the blood circulatory system via connections of the thoracic duct to the internal jugular vein.
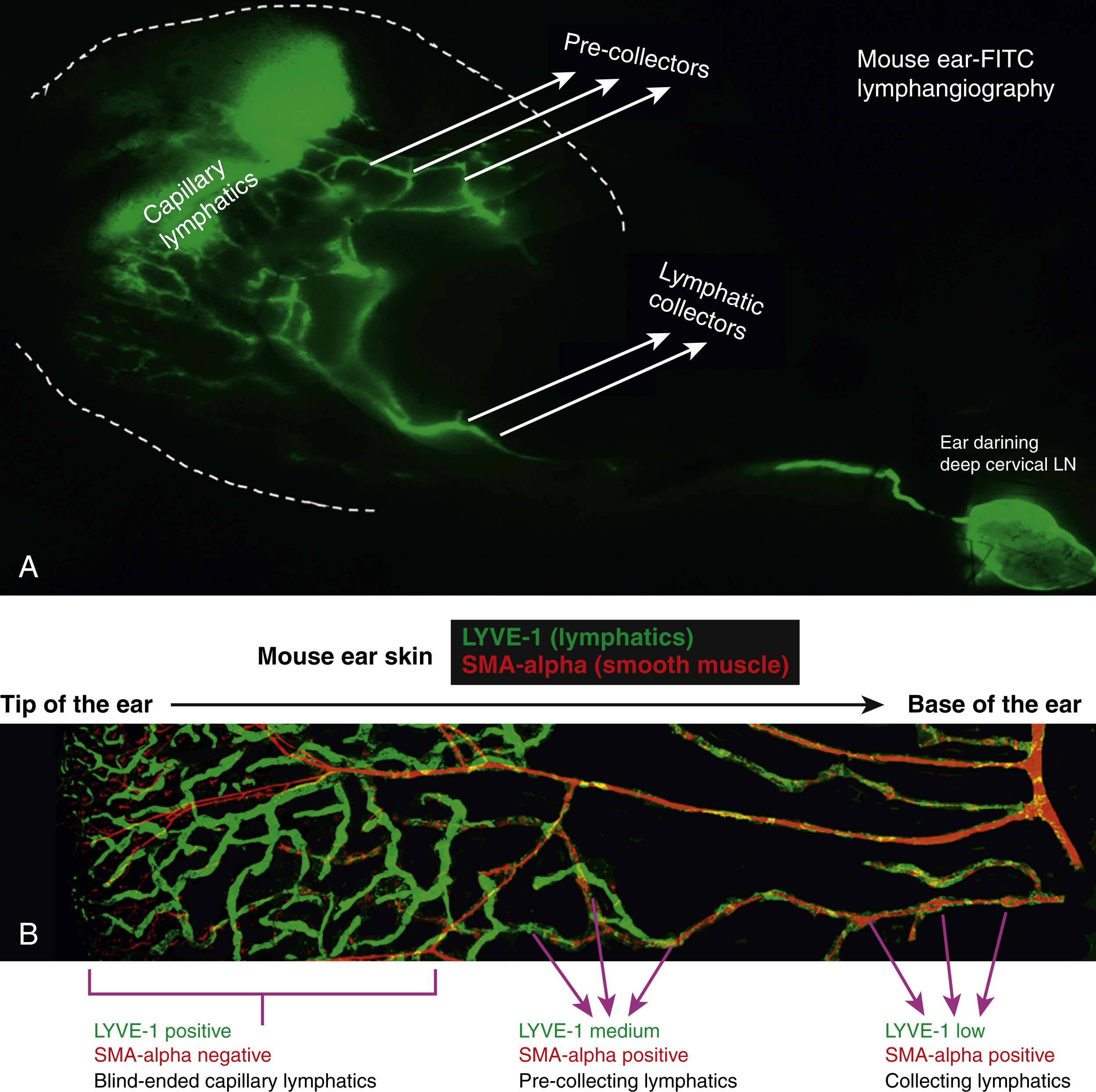
The capillary lymphatics, located in the dermis, have a single layer of lymphatic endothelia cells connected by overlapping or intercalating button-like junctions. These cells are tethered to the surrounding tissue by anchoring filaments, which pull open the button-like junctions between individual cells and widen the lymphatic vessel lumen as interstitial volume increases. This allows fluid and molecules to enter the lymphatic system. Once in the system, fluid and particulate matter reach the collecting lymphatics. These collecting lymphatics are comprised of continuous or zipper-like endothelial cell junctions that prevent fluid leak, a surrounding basement membrane and smooth muscle cells that coordinately contract to propel fluid forward, and bicuspid valves that ensure one-way flow. Passive forces such as muscle contractions, respiration, regional arterial pulsation, and gravity also assist in moving lymphatic fluid within collecting vessels.
The lymphatic system is composed of superficial and deep collecting lymphatic vessels, divided by the deep fascia. The superficial and deep systems have no direct connections between them, although lymphatic vessels do diverge and converge within each system. The lymphatic drainage of the skin can be divided into lymphatic territories called lymphosomes. Anatomical dissections in canine specimens by Suami and colleagues, and more recently indocyanine green lymphography in the clinical setting, have revealed multiple lymphosomes draining the skin. In addition, these studies have shown that collateral lymphatics may develop following injury bypassing the area of lymphatic obstruction. This observation is important and may provide an anatomical rationale for the finding that lymph node dissection does not result in the development of lymphedema in all patients.
In the upper extremity, lymphatic collectors emerging from the fingers and palm run axially in the dorsal hand and divide into radial and ulnar bundles. These collecting lymphatic vessels travel up the forearm, converging towards each other, and gradually changing course at the elbow to the anteromedial aspect of the limb. The primary drainage pathway of the upper extremity drains along the medial aspect of the upper extremity, within the bicipital groove, eventually draining into the axillary region ( Fig. 44.2 ). An alternate pathway in some individuals extends from the dorsal surface of the lateral upper arm and drains into the supraclavicular lymph nodes, bypassing the axillary lymph nodes. This pathway can develop as a collateral drainage route in some patients who undergo axillary lymph node dissection.
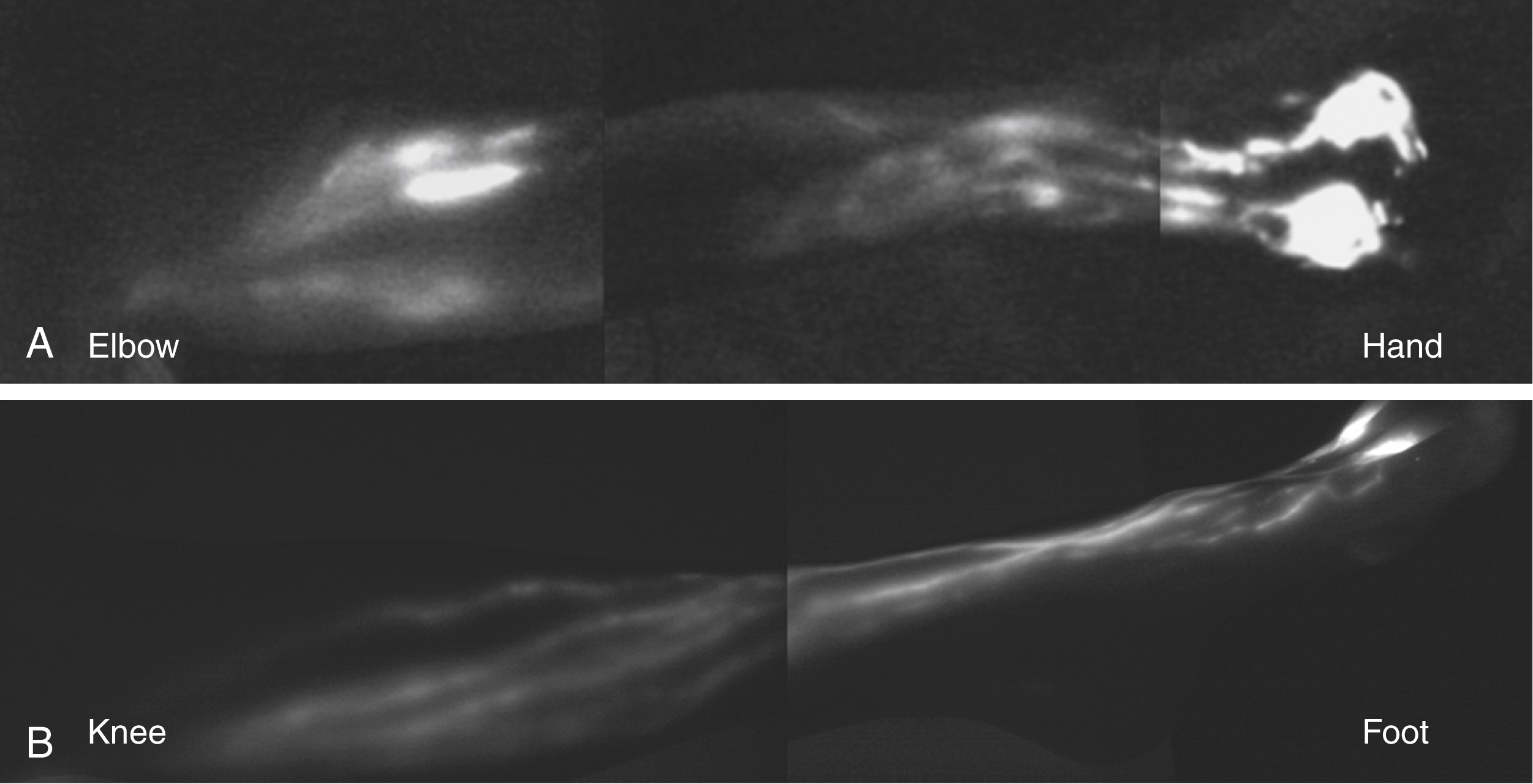
In the lower extremity, lymphatics emerging from the toes and soles of the feet travel from the medial and anterior foot, ascending axially along the medial border of the leg (see Fig. 44.2 ). The vessels in the lower extremity range in size from 0.2 mm to 2.2mm in diameter and travel in the subcutaneous tissues. These lymphatic channels have a tortuous course and drain along the lesser saphenous vein, draining into the popliteal lymph nodes. Other collectors bypass the popliteal region and drain along the medial aspect of the thigh along the greater saphenous vein, draining into the inguinal lymph nodes.
Lymph Nodes
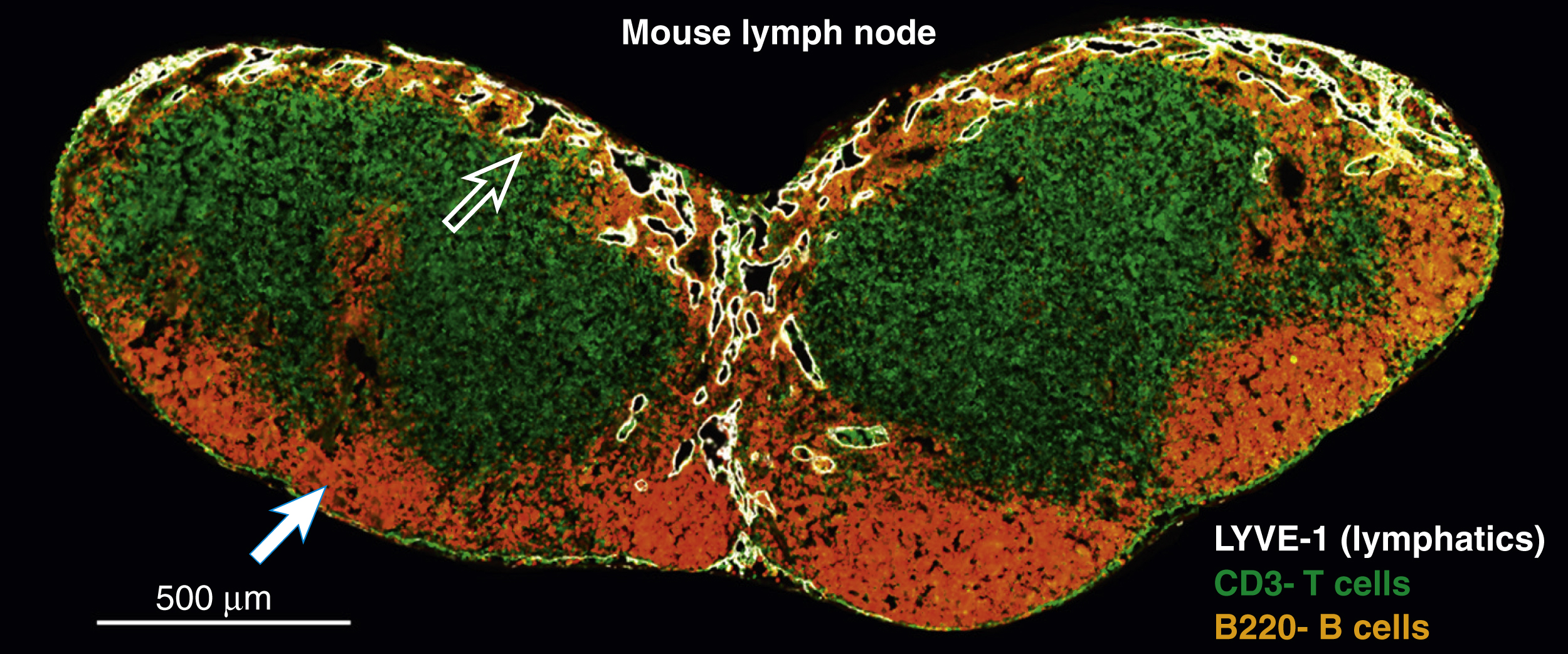
Lymphatic fluid is filtered by lymph nodes to allow sampling of bacterial antigens, removal of particulate matter, and transport of antigen-presenting cells to areas where T and B cell activation can occur. Lymph nodes are localized in the abdominal cavity as well as at the junction of the extremities and the head with the trunk. Interstitial fluid in the afferent lymphatics drains into the subcapsular sinus of the lymph node and through lymphatic sinuses surrounding B cell follicles ( Fig. 44.3 ). Lymphatic sinuses are lined by reticular endothelial cells and dendritic cells that are activated by antigens within the lymphatic fluid. Particulate matter and bacteria are phagocytosed by lymph node-resident macrophages that process the antigens and promote activation of immune responses. Immune cells from the systemic circulation enter and leave the lymph node through high endothelial venules that border lymphatic sinuses, thus enabling the exchange of cells, and to a lesser extent, interstitial fluid with the systemic circulation. The lymphatic fluid continues to drain to the medulla (central portion) of the lymph node and exits the node via efferent lymphatics that continue to drain into the remaining lymph node chains and eventually into the thoracic duct to empty into the venous system.
Etiology of Lymphedema
Primary Lymphedema
Recent advances in DNA sequencing have identified a variety of genetic abnormalities that are associated with the development of primary lymphedema. These abnormalities involve a broad spectrum of pathologic changes, including abnormal lymphangiogenic growth factor signaling, lymphatic valve defects, leaky tight junctions, smooth muscle cells coverage, and alterations in lymphatic contractility. The most common cause of primary lymphedema is an insufficient number of collecting lymphatics. These genetic defects may be familial or may develop spontaneously as a result of germline or somatic mutations.
In most cases of primary lymphedema, the genetic defects that underly the development of the disease remain unknown. Nevertheless, it is generally thought that lymphatic abnormalities in these cases are present at birth and gradually worsen with time.
The presentation of familial forms of lymphedema is highly variable both in terms of timing and severity of symptoms. These differences may be related, at least in part, to the penetrance of the genetic abnormalities. Other causes of differences in severity or timing of lymphedema presentation include interaction with environmental factors (e.g., obesity) or hormonal fluctuations. As a result, some cases of primary lymphedema referred to as congenital lymphedema , present early in life or shortly after birth. More commonly, primary lymphedema becomes manifest later in life, a condition termed lymphedema praecox or lymphedema tarda .
Congenital lymphedemas account for 10%–25% of all cases of primary lymphedema and occur most commonly in the lower extremity of females. The severity of this disease is highly variable, ranging from mild to severe swelling. In addition, congenital lymphedema may occur unilaterally or differentially between limbs. Milroy’s disease is a classic form of congenital lymphedema and accounts for 2%–3% of all patients with this form of lymphedema. Patients with this familial, sex-linked disease have heterozygous inactivating mutations of vascular endothelial growth factor 3 (VEGFR3) and usually, present shortly after birth, with limb swelling and/or chylothorax. Abnormalities in VEGFR3 signaling are important because activation of this receptor by its ligands, vascular endothelial growth factor C (VEGF-C) and the closely related VEGF-D, is necessary for lymphatic endothelial cell development, proliferation, differentiation, and migration. As a result, patients with Milroy’s disease have hypoplastic lymphatic vessels. Interestingly, however, the severity of lymphatic vessel abnormalities can vary between the upper and lower extremities (lower extremities are more severely affected) and abnormalities are often absent in intestinal lymphatics.
Patients diagnosed with lymphedema praecox present with lymphedema before the age of 35. Most patients diagnosed with lymphedema praecox are female (ratio of 4:1 with males) and develop unilateral lower extremity lymphedema at or around the time of puberty, suggesting that sex hormones may contribute to the development of this disease. Although pathological changes in the lymphatic system in patients with lymphedema praecox are highly variable, most patients display decreased numbers of initial lymphatics and hypoplastic collecting vessels.
Patients who develop primary lymphedema after the age of 35 have lymphedema tarda ( Fig. 44.4 ). This is a diagnosis of exclusion and other causes of lymphedema or diseases that may cause swelling must be ruled out. This presentation of primary lymphedema is also infrequent and, like other forms of primary lymphedema, occurs most commonly in the lower extremity of women. Recent genetic studies have suggested that some patients with lymphedema tarda have mutations in FOXC2 , a gene that regulates lymphatic valve development and function.
An arbitrary classification of primary lymphedema based on the age at which symptoms present is an oversimplification and most likely not helpful when evaluating individual patients. These schemes do not, for example, provide any basic knowledge of the pathophysiology of the disease and, as a result, do not help in devising logical treatment options. More recent studies have developed classification schemes that combine presentation of the disease and genetic mutations. This approach categorizes congenital lymphedemas into five main groups: syndromic, systemic or visceral, disturbed growth, congenital onset, and late onset. This classification is more precise and may be more useful than the timing of presentation alone in developing diagnostic or therapeutic interventions.
Secondary Lymphedema
Secondary lymphedema results from obstruction or injury to the lymphatic system. Worldwide, the most common cause of lymphedema is infection by roundworms ( Wuchereria bancrofti , Brugia malayi , and Brugia timori ). The roundworms, commonly transmitted by mosquitos, grow in the tissues and release larvae that occlude lymphatic channels. This occlusion, and the resultant severe inflammatory response, cause progressive and often devastating limb swelling. Treatment for filariasis is symptomatic and primarily limited to antiparasitic medications that kill the developing larvae rather than the adult worm residing in the tissues. Patients with severe filariasis-induced lymphedema often require disfiguring reductive surgery.
In the developed world, the most common cause of secondary lymphedema is iatrogenic injury to the lymphatic system in the course of cancer treatment. Lymphedema can occur from the treatment of all solid tumors, including breast, gynecological tumors, melanoma, sarcoma, pelvic tumors, and even head and neck tumors. Although the rates of and severity of the disease vary depending on the extent of the surgery and adjuvant therapies (e.g., radiation), most patients who undergo lymph node dissection or even wide skin excision are at risk for developing lymphedema.
The development of lymphedema after surgery is usually delayed and occurs 3–12 months postoperatively. Thus, while most patients have transient postoperative swelling immediately after surgery, these changes typically resolve only to recur in a progressive manner in a subset of patients who go on to develop lymphedema. Lymphedema tends to develop more rapidly in the lower extremities as compared with the upper extremities. Thus, patients who undergo pelvic surgery and develop lymphedema typically do so within a few months of surgery. In contrast, patients treated for breast cancer usually develop lymphedema 8–10 months following surgery. Nearly 75% of patients who develop lymphedema after axillary lymph node dissection for breast cancer do so within the first 3 years following surgery. However, lymphedema develop even 30 years after the initial surgical insult has been reported.
Progression of lymphedema, once it develops, is highly variable with some patients displaying slowly progressive disease, even without compression, while others exhibit a more fulminant form of the disease with rapidly progressive symptoms despite aggressive therapy. Nevertheless, early diagnosis and aggressive physical therapy/compression is helpful in most patients and should be instituted as soon as possible as early intervention has been shown to improve long-term outcomes. Other nonsurgical interventions, for example, exercise or weight loss programs, can also be helpful in preventing the development of disease or decreasing the rate of progression once it has developed. The concept that exercise or weight lifting can increase “lymphatic load” has been disproven by several level 1 randomized prospective control studies demonstrating the efficacy of these interventions for the treatment of lymphedema. Thus, patients who are at risk for developing lymphedema, as well as those who have developed lymphedema, should be encouraged to maintain a healthy weight and participate in aerobic and resistance exercise.
Risk Factors for Lymphedema
There are many known risk factors for the development of lymphedema, including obesity, radiation, infection, advanced age, and genetic factors. Obesity has long been recognized as an important risk factor for the development of lymphedema following cancer surgery. For example, Helyer et al found that patients with a body mass index (BMI) of greater than 30 had a more than threefold increase in the risk of developing lymphedema following breast cancer treatment as compared with patients with a BMI less than 25. McLaughlin et al examined breast cancer patients treated with either sentinel lymph node biopsy or axillary lymph node dissection and found those with a higher baseline BMI had a significantly higher incidence of lymphedema. Interestingly, these authors found that even postoperative weight gain (independent of the presurgical weight) significantly increased the risk of lymphedema development. Importantly, a randomized, controlled, prospective trial showed that patients who had nutritional counseling and lost weight over a 12-week period of time had significant reductions in their upper extremity limb volumes as compared to a control group that did not receive nutritional counseling and did not lose weight. Other studies have shown that exercise programs improve lymphedema symptoms at least in part by promoting weight loss. These findings are supported by prospective longitudinal studies demonstrating that the incidence of lymphedema is increased nearly twofold in patients with a sedentary lifestyle.
Several lines of evidence support the idea that obesity may increase the risk of lymphedema by decreasing lymphatic function. For example, macromolecule transport in obese patients is significantly decreased as compared with healthy volunteers. Other studies have shown that obese patients (BMI >59) spontaneously develop lower extremity lymphedema even without surgery or other injuries. Preclinical studies have shown that increasing obesity is linearly correlated with impaired lymphatic pumping capacity, lymphatic vessel number, and lymphatic vessel leakiness. These changes are partially reversible with exercise or weight loss. Taken together, these findings suggest that weight loss and exercise programs may be beneficial in patients who are considered for lymphatic surgery since these behavioral modifications improve lymphatic function and regeneration.
Radiation therapy , mainly when used as adjuvant therapy in conjunction with surgical extirpation, is also a significant risk factor for the development of lymphedema increasing the relative risk of disease two- to fivefold. Radiation in isolation rarely (<7%) results in lymphedema development. However, radiation and obesity appear to increase the risk of lymphedema in an additive manner since the risk of disease development is significantly higher in obese patients treated with adjuvant radiotherapy. Some researchers have questioned the link between radiation and development of breast cancer-related lymphedema, having failed to show a significant increase in risk in some studies. However, it is likely that these studies are confounded by the fields used for radiation and heterogeneous populations of patients that had radiation treatment to the chest wall rather than regional lymph node basins.
Postoperative infections and cellulitis have also been linked to the development of lymphedema following lymph node dissection for a variety of cancers. This clinical finding is supported by recent studies demonstrating that bacterial toxins and inflammatory responses can permanently injure lymphatic endothelial cells and lymphatic smooth muscle cells. Other studies have shown that lymphatic injury can result in impaired innate and adaptive immune responses, thereby increasing the risk of infections. Thus, patients with lymphatic injury are at high risk for developing infections and when these infections develop, they may cause additional injury to the lymphatic system, thereby setting up a vicious cycle that leads to rapid progression of the disease.
Advanced age also infers increased risk of developing lymphedema in some series. One study of 287 Australian breast cancer survivors showed that women who were more than 50 years old were 3.1 times more likely to develop lymphedema following axillary lymph node dissection as compared with those who were less than 50 years old at the time of surgery. However, the importance of age as a predictive factor for lymphedema development is debated, as other studies have failed to show a significant correlation. Nevertheless, recent preclinical studies have shown that aging decreases lymphatic pumping capacity, lymphatic permeability, and lymphatic endothelial cell proliferation potential with resultant decreased lymphatic function. It is therefore conceivable that these physiological changes may infer a higher risk of developing lymphedema after surgery.
Recent studies have identified genetic mutations and alternative coding regions that increase the risk of secondary lymphedema development. For example, Finegold et al showed that mutations in the connexin 47 or hepatocyte growth factor genes significantly increased the risk of breast cancer-related lymphedema. Other studies have shown that normal variants of four genes, so-called single-nucleotide polymorphisms (SNPs), confer a significant risk for the development of secondary lymphedema following axillary lymph node dissection for breast cancer treatment. A recent meta-analysis reviewed reports of the association of secondary lymphedema and genetic variations in coding sequences of 18 genes, including HGF , MET , GJC2 , IL1A , IL4 , IL6 , IL10 , IL13 , VEGF-C , NFKB2 , LCP-2 , NRP-2 , SYK , VCAM1 , FOXC2 , VEGFR2 , VEGFR3 , and RORC . Taken together, these studies demonstrate that lymphedema is a complex disease and that the risk of this disease can be modified by intrinsic and extrinsic risk factors. It is also clear that these independent variables interact with each other, further modulating the risk of disease development.
Pathophysiology of Secondary Lymphedema
Recent studies have helped elucidate the pathophysiology of secondary lymphedema and provided some answers to vexing questions about this disease. To understand these discoveries, it is essential to have a historical perspective of clinical and experimental studies aiming to understand this disease.
For many years, it was assumed that the pathophysiology of secondary lymphedema is related to lymphatic injury and the failure of lymphatics to regenerate or form collateral pathways that bypass the zone of injury. This failure of regeneration resulted in a mismatch between lymphatic transport capacity and interstitial fluid production, leading to the development of lymphedema. Indeed, this hypothesis was supported by laboratory studies demonstrating that delivery of lymphangiogenic growth factors such as VEGF-C improved lymphatic regeneration and resulted in resolution of lymphedema in animal models. The discovery of associations between coding variants or mutations in genes regulating lymphatic vessel development or differentiation in a small subset of patients who developed secondary lymphedema also supported this hypothesis. However, more recent studies demonstrated that the delivery of lymphangiogenic growth factors does not improve lymphedema, and in some circumstances, the expression of these factors (including VEGF-C) is paradoxically increased in lymphedematous tissues harvested from clinical biopsy specimens. More importantly, the lymphatic injury hypothesis failed to provide a rationale for the clinical characteristics of the disease. For example, lymphatic injury alone does not explain why lymphedema develops in a delayed manner, usually months and sometimes years after the initial surgical insult. If lymphatic damage is the cause of lymphedema, then why do some patients develop the disease while others do not? Why do some patients with seemingly minor lymphatic injury (e.g., sentinel lymph node biopsy) develop lymphedema? Why is lymphedema progressive, and why is the rate of progression so highly variable? These characteristics of secondary lymphedema suggested that lymphatic injury is an initiating event and that in some patients additional pathways are activated, leading to disease development and progression.
Lymphedema is a progressive disease of the entire lymphatic vascular tree rather than an isolated injury at the site of lymph node dissection. This concept is supported by progressive changes noted in indocyanine green (ICG) lymphography demonstrating incremental increases in lymphatic vascular leakiness (as evidenced by dermal backflow), loss of collecting lymphatics, and formation of aberrant lymphatic channels to bypass lymphatic obstruction at multiple levels of the limb. The end-stage of the disease is characterized by loss of all functional lymphatics and pooling of lymphatic-specific dyes in the skin. These changes have led to the development of ICG lymphedema classification systems (see below).
Histologically, lymphedema is characterized by progressive accumulation of fibroadipose tissues in the skin. This fibrotic process also involves the lymphatic vessels encasing the initial and collecting vessels in thick collagen bundles. As lymphedema progresses, collecting lymphatics become progressively sclerosed with the proliferation of lymphatic smooth muscle cells and obliteration of the vessel lumen and resultant lymphatic dysfunction. Although the mechanisms that regulate the proliferation of smooth muscle cells remain unknown, it is hypothesized that increased afterload resulting from the initial surgical insult (and tissue fibrosis) may, in a fashion like hypertensive cardiomyopathy, lead to a compensatory increase in the number of smooth muscle cells. Progressive fibrosis and lymphatic vessel obliteration provide a rationale for the finding that lymphedema develops in a delayed fashion after surgery since these changes take time to happen. In this manner, lymphedema may simply be fibrotic organ failure of the lymphatic system. This hypothesis is supported by the fact that fibrosis is a common end-pathway for organ failure in all other organ systems. In these diseases, similar to lymphedema, the progression of the disease is often highly variable and influenced by genetic or environmental factors. For example, obesity, recurrent infections, and radiation (i.e., important risk factors for lymphedema) are also essential regulators of fibrosis in a variety of other fibroproliferative disorders. Thus, it is possible that surgical injury to the lymphatic system in some patients initiates progressive changes in the tissues/lymphatics of the limb that then lead to the development of lymphedema in some individuals if a critical threshold of decreased lymphatic function is reached. Patients with risk factors for fibrosis such as obesity, infections, radiation, genetic abnormalities, advanced age, etc. are more likely to reach this threshold because these risk factors independently increase the risk of fibrosis.
Fibrosis in lymphedema is regulated by chronic inflammatory responses that involve T-helper cells. These cells produce profibrotic growth factors such as interleukin 4 (IL4), IL13, and transforming growth factor beta-1 (TGF-β1) that promote collagen deposition, decrease matrix breakdown, and regulate fibroblast differentiation. These T cell inflammatory reactions also regulate lymphatic pumping, leakiness, and formation of collateral pathways suggesting that T cells play a key role in the pathophysiology of lymphedema.
The discovery that inflammation is a crucial regulator of lymphedema development led to studies aiming to determine the efficacy of antiinflammatory drugs in lymphedema treatment. An early preclinical study in this area showed that ketoprofen, a nonsteroidal antiinflammatory drug, was effective in decreasing the severity of lymphedema in mouse models. This finding led to a clinical trial of ketoprofen with 55 patients. Interestingly, although the authors failed to show significant improvements in limb volumes (the primary endpoint of the study), they did note significant improvements in skin fibrosis and thickness. Subsequent studies have shown that the beneficial effects of ketoprofen are primarily related to activation of leukotriene B4 and have led to the initiation of new clinical trials using more targeted approaches.
Other studies have more directly targeted T cells for the treatment of lymphedema. For example, a recent study by our lab showed that topical application of tacrolimus, a drug that decreases T cell proliferation and differentiation, could prevent the development of lymphedema in a mouse model of the disease. This treatment was also highly effective in this mouse model for treating established lymphedema. These findings suggest that inflammatory pathways activated by lymphatic injury play a key role in the development of lymphedema. More importantly, these studies continue to identify novel means of treating lymphedema. These treatments may be even more effective when combined with surgery to prevent the development of lymphedema or treat it once it has developed. This is an exciting new area in lymphedema research.
Lymphedema Diagnosis
Lymphedema is diagnosed by history, physical exam, and objective measures. Symptoms of lymphedema include gross or perceived swelling of the limb, pain, decreased range of motion, subjective sensation of heaviness, and tightness. These symptoms can significantly impair quality of life and limit the ability of affected individuals to participate in activities of daily living. Some patients also have a history of recurrent infections that often require intravenous antibiotic treatment. A history of trauma, surgery, or radiation as well as family history, should also be elicited. The history should also include a detailed analysis of the onset and course of lymphedema and the treatments that the patient has had for the condition, including physical therapy, compression, and manual lymphatic massage.
Physical Examination
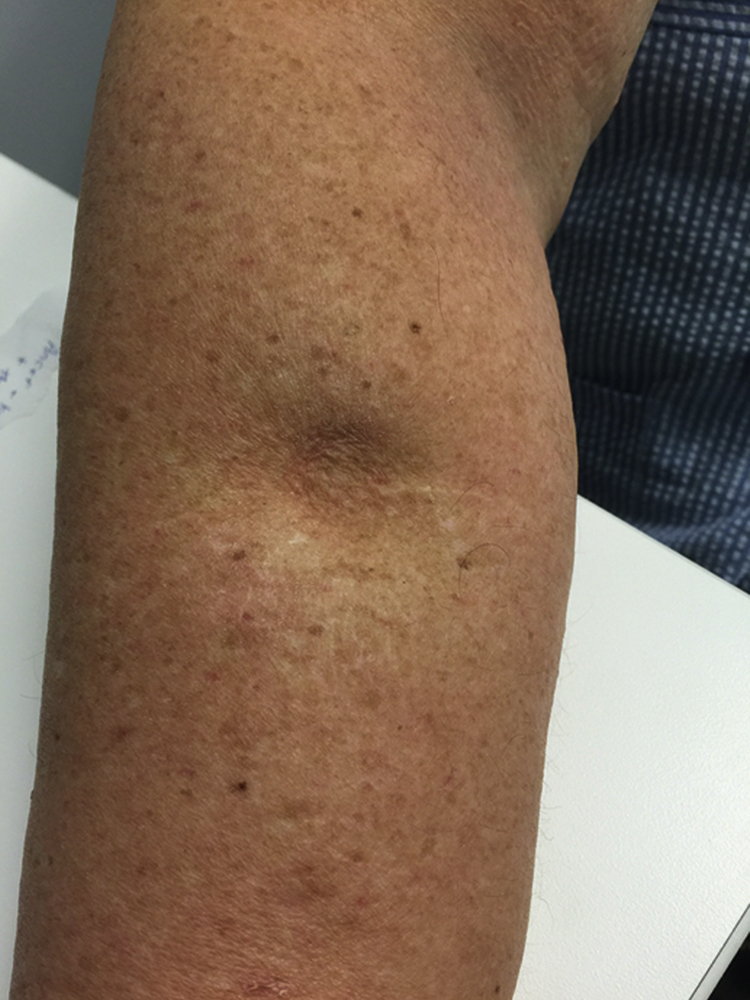
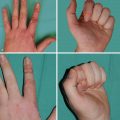
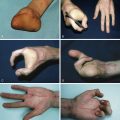
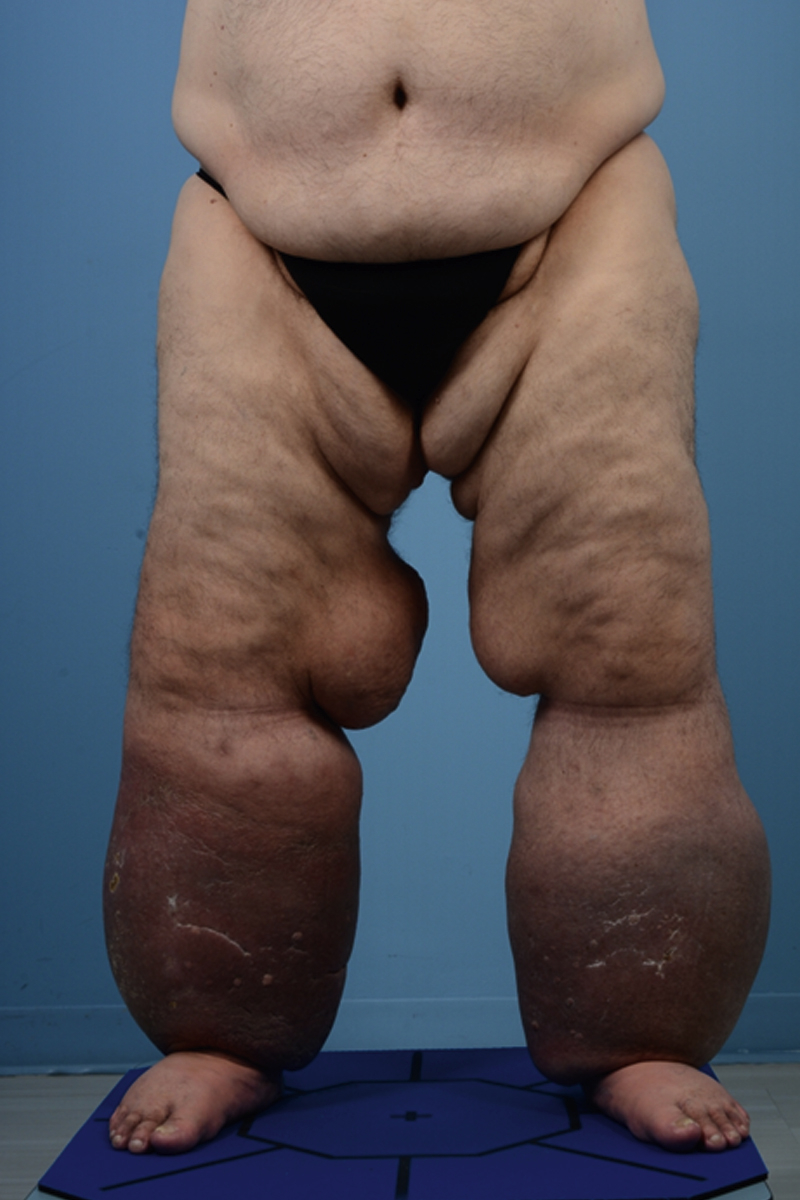
Physical exam findings in patients with lymphedema are variable and can range from a healthy limb to an enlarged limb with pitting edema, to an enlarged limb with thickened, fibrotic, and nonpitting skin. Swelling can be diffuse or focused in some regions of the limb. The dorsal aspect of the arm just distal to the elbow and the lateral/dorsal aspect of the upper arm are frequently involved ( Fig. 44.5 ). Less common, though more problematic, is hand or finger swelling. The differential diagnosis of lymphedema includes infection, venous insufficiency, recurrent malignancy with resultant venous or lymphatic obstruction, and, less frequently, heart failure.