Lyme Borreliosis: Introduction
|
Lyme borreliosis, or Lyme disease, is the most commonly reported arthropod-borne illness in both the United States and Europe.1 It was first recognized in the 1970s after epidemiologic investigations of a clustering of cases of oligoarthritis among children in eastern Connecticut established a probable microbial etiology for the disease.2 In 1981, Burgdorfer isolated a new spirochetal bacterium, Borrelia burgdorferi, from the midgut of the Ixodes dammini tick (now Ixodes scapularis).3 Recovery of the organism subsequently from cutaneous lesions, cerebrospinal fluid (CSF), and blood specimens of patients with Lyme disease in both the United States4,5 and Europe6–8 definitively linked the disease with B. burgdorferi.
Epidemiology
The existence of B. burgdorferi in the Northeastern United States likely predates the presence of European settlers by several thousand years.9 The incidence and range of the organism has steadily grown since its recognition in the 1970s. Deer and birds are thought to be the primary drivers in dispersal of infected ticks into new areas. In the United States, the Centers for Disease Control and Prevention (CDC) initiated surveillance for Lyme disease in 1982, and the Council of State and Territorial Epidemiologists made Lyme disease a nationally notifiable disease in 1991. Since 1992, 248,074 cases have been reported to the CDC.10 In 2006, the number of new cases in the United States was 19,931 (national median incidence 0.5 cases per 100,000 people). This represents a 101% increase in annual incidence since 1992. There is believed to be significant underreporting of Lyme disease, and the real number of new cases each year in the United States is actually thought to approach 150,000. For the 15-year period for which the CDC has data, approximately 93% of reported cases occurred in ten states located in the Northeastern, mid-Atlantic, and North Central regions: Connecticut, Delaware, Maine, Maryland, Massachusetts, Minnesota, New Hampshire, New Jersey, New York, Pennsylvania, Rhode Island, and Wisconsin. Nearly all states have reported cases of Lyme disease at some point during the past 15 years, though average incidence varies markedly from 0.0 for Colorado, Montana, and Hawaii to 73.6 per 100,000 people for Connecticut. Disease in many of the states with low incidence represents travelers infected during travel to more endemic regions.
Lyme disease is also widely distributed in Europe, with an estimated 120,000 new cases each year.11 The highest reported frequencies occur in forested areas of central and Northern Europe (Germany, Austria, Slovenia, and Sweden).12,13 The infection is also found in Eastern Russia, China, Korea, and Japan.14
Etiology
Borrelia burgdorferi sensu lato, the agent of Lyme borreliosis, belongs to the eubacterial phylum of Spirochaetales, which are vigorously motile, corkscrew-shaped bacteria. The borrelial genome is made up of a linear chromosome and more than 20 circular and linear plasmids, the largest number known for any bacterium.15 Some of the plasmids can be considered minichromosomes, as they are required for survival of the organism. Among the interesting characteristics of the organism are the large number of lipoproteins (more than 150). B. burgdorferi‘s genome encodes no toxins or other identified virulence factor. Borrelia also lack biosynthetic machinery to produce many essential nutrients (e.g., amino acids and fatty acids) suggesting that B. burgdorferi is highly dependent on its host for obtaining crucial nutrients.15
Eleven Borrelia spp. have been described worldwide within the B. burgdorferi sensu lato family. Only three—(1) B. burgdorferi sensu stricto, (2) Borrelia afzelii, and (3) Borrelia garinii—have been confirmed to be pathogenic in humans and to cause Lyme borreliosis, although other species are under investigation for their relationship to human diseases (including Borrelia bissettii16,17 and Borrelia lonestari18).
In the United States, the etiologic agent of Lyme disease is exclusively B. burgdorferi sensu stricto. B. afzelii and B. garinii cause the majority of Lyme disease in Europe, although B. burgdorferi sensu stricto is also present.19 In Asia, only B. afzelii and B. garinii have been confirmed to exist.
B. burgdorferi is maintained in nature through a cycle involving small rodents, birds, and ticks of the Ixodes species.20,21 Although not important reservoirs for B. burgdorferi, deer are an important feeding source for adult ticks and reductions in deer populations can lead to significant decreases in the tick population.22
Ixodes ticks have a 2-year, three-stage (larval, nymphal, and adult) life cycle. Larval ticks acquire B. burgdorferi organisms by taking a blood meal from an infected animal and maintain the infection during the subsequent molting to the nymphal and adult stages. B. burgdorferi remain dormant in the tick’s midgut between feedings.23 Each tick life stage takes one blood meal: larval ticks feed in late summer, nymphal ticks feed the subsequent spring and early summer, and adult ticks feed in the fall and early winter. The major reservoirs for B. burgdorferi are small rodents and birds that are fed on by larval and nymphal ticks. Adult ticks feed on larger mammals (e.g., deer) that are not important reservoirs for B. burgdorferi. Humans are incidental hosts not important in maintaining B. burgdorferi in the wild. Most cases of human illness occur in the late spring and summer months when the ticks are most active and human outdoor activity is greatest.
Of clinical relevance, certain Ixodes ticks are vectors of other tick-borne illnesses in addition to Lyme disease: I. scapularis ticks in the United States and Ixodes ricinus ticks in Europe may transmit Babesia microti (a red blood cell parasite) or Anaplasma phagocytophilum (formerly referred to as the agent of human granulocytic ehrlichiosis).24,25 I. ricinus in Europe and Ixodes persulcatus in Asia are also vectors of tick-borne encephalitis virus.26
Once in the host’s skin, B. burgdorferi begins to spread from the initial inoculation site rapidly into the bloodstream where it disseminates to multiple sites. Time from inoculation to dissemination varies from as little as 3 days to weeks.5
B. burgdorferi has developed strategies to adapt to and survive in markedly different microenvironments.27 The organism expresses a variety of proteins during its life cycle, facilitating binding to different host proteins, migration within host tissues, transmission, and evasion of the host immune response.28,29 A well-documented example of host adaptation is the variable expression of outer-surface proteins (Osp) A and C. OspA, primarily expressed while the spirochete is dormant in the tick’s midgut,30 is thought to serve as an anchor by binding the tick midgut protein, TROSPA.31 When the tick engorges on a host, B. burgdorferi downregulates OspA, releases from the midgut, and begins to multiply and travel to the salivary glands. This process takes between 24 and 48 hours, which explains why removal of the tick before 24 hours of attachment prevents transmission of disease.32
As the organism downregulates OspA, it upregulates OspC, which facilitates migration from the tick’s gut to its salivary glands. OspC binds to a tick salivary protein, Salp15, which protects the organism from clearance by host immune defenses.33 Immunosuppressive characteristics of tick saliva also aid in the establishment of early infection. For example, tick saliva contains proteins that bind host chemokines.34 These proteins, termed evasins, help suppress recruitment of inflammatory cells into the area where the tick is feeding allowing the tick to better feed and incidentally providing protection to organisms such as B. burgdorferi that are transmitted during feeding.
B. burgdorferi also bind host factor H to protect itself from complement-mediated killing.35,36 Specificity of strains of B. burgdorferi for factor H from different animal species may, in part, explain differences in host range between the species. B. burgdorferi have been shown to upregulate host proteases such as matrix metalloproteinases in the skin and joints which may play a role in pathogenesis by digesting extracellular matrix proteins and allowing dissemination, but this requires further clarification.37
Both innate and acquired host immune defenses have important roles in the control of B. burgdorferi infection; however, neither can typically eradicate the disease.38,39 Phagocytosis, both complement mediated and noncomplement mediated, is the first line of host defense against B. burgdorferi.40,41 Natural antibodies (polyclonal IgM antibodies) may also function as a first line of defense against B. burgdorferi, killing spirochetes in the tick midgut during the blood meal and limiting pathogen burden before the development of a specific adaptive immune response.
Once inside the host, B. burgdorferi is a strong inducer of inflammation. Its outer surface lipoproteins are potent inflammatory stimuli.42–44 Recognition of borrelial lipoproteins occurs through binding to Toll-like receptor 1 and 2 heterodimers, which initiates a cascade of intracellular signaling events culminating in the production of proinflammatory cytokines, chemokines, and other molecules important in host defense. Studies using Toll-like receptor-deficient mice have shown that other signaling pathways are also activated by B. burgdorferi and result in release of inflammatory mediators. Among the other innate immune receptors recognizing B. burgdorferi and mediating inflammation are integrin receptors.45,46
Development of a specific humoral immune response generally occurs over weeks to months.47,48 The IgM response usually peaks between 3 and 6 weeks after disease onset and may remain elevated for months to years; the immunoglobulin G (IgG) response usually follows the IgM response by several weeks so that approximately 90% of patients have detectable IgG levels 4–6 weeks into the infection. The development of the humoral antibody response typically heralds a significant decrease in the number of organisms and decrease in the level of inflammation. Of note, the acquired humoral response is not typically protective against reinfection, in part due to significant sequence variability in protective antigenic epitopes between B. burgdorferi strains and the lack of expression of less variable protective antigens (e.g., OspA) in the mammalian host.49
Clinical Manifestations
The clinical course of Lyme disease is typically described in stages that parallel that of another spirochetal disease, syphilis: early localized, early disseminated, and late disseminated. Localized disease closely follows the bite of an infected tick and is characterized by the erythema migrans (EM) rash with or without constitutional symptoms. Early disseminated Lyme disease presents days to months after onset of infection as a number of distinct clinical entities affecting the skin, joints, nervous system, and heart. Late-disseminated Lyme disease typically presents in only a subgroup of untreated patients months to years later, and manifests as chronic symptoms localized most often to the skin, joints, and nervous system. A minority of patients go on to develop prolonged posttreatment syndromes. These include a syndrome of antibiotic-resistant arthritis thought possibly to be autoimmune in nature, and, separately, a constellation of symptoms with similarities to fibromyalgia and chronic fatigue syndrome. The relationship of these latter symptoms to infection with B. burgdorferi is still debatable.
The disease caused by each species of B. burgdorferi is largely the same but certain clinical manifestations are more closely associated with infection due to a particular species.19,50 For instance, B. burgdorferi sensu stricto is the most arthritogenic of the three Borrelia species.51 For this reason, Lyme arthritis is the most frequent presentation of late Lyme disease in North American patients. Similarly, because B. afzelii is the species that has most often been associated with late skin manifestations; some dermatologic conditions [e.g., acrodermatitis chronica atrophicans (ACA) and borrelial lymphocytoma] are nearly exclusively found in Europe. Finally, B. garinii, the most neurotropic of the three species, causes a wide range of neurologic abnormalities.
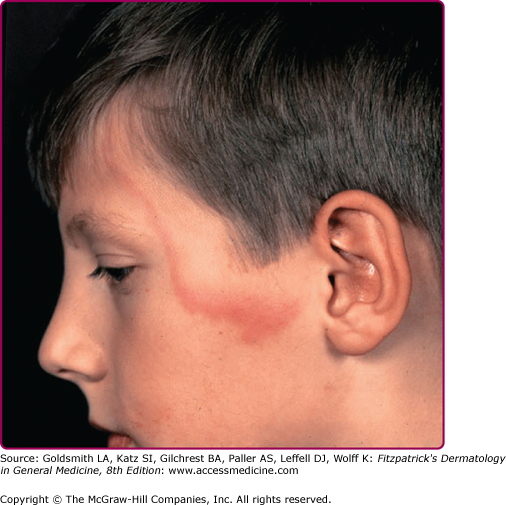
In early case reports, EM lesions were reported in 50%–80% of patients with Lyme disease. More recent studies have shown lower rates of EM, likely due to improved patient education. Definite history of tick bite at the site of the lesion is obtained in only a small proportion of patients.52 The lesion itself is believed to be the result of the direct presence of the spirochete, corroborated by reports showing aspiration and cultivation of the organism from the lesion (see Fig. 187-1). EM lesions develop within 3–30 days of the tick bite (median, 7 days).53
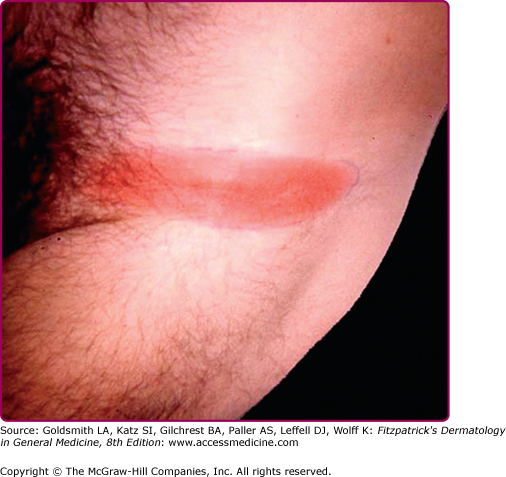
EM may be seen anywhere on the body but is most common on the lower extremities, inguinal and axillary regions of adults (Fig. 187-2), and on the face in children (“slapped cheek” appearance) reflecting sites of predilection of the tick.2 Although no definite sex predilection has been noted in the United States, women are reported to be more commonly affected in European studies.8
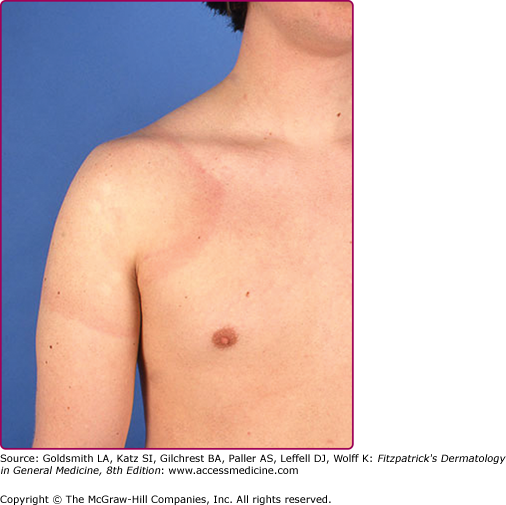
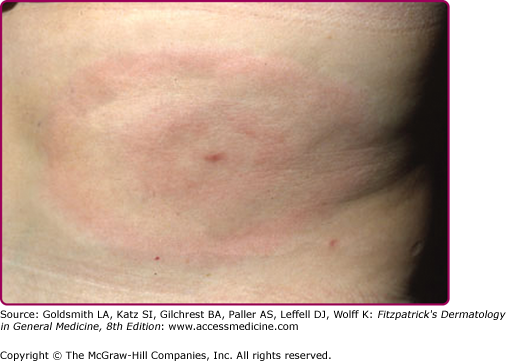
The skin lesion has a characteristic and pathognomonic appearance (see Fig. 187-2) of an expanding erythema encircling the bite site, with the transition between the central zone and periphery being less well demarcated than between that of the periphery and adjacent skin.52 The border is usually continuous and not patchy. Cases in which the erythema appears stationary or even linear are reported.5 Typically described as round, the lesion in reality is more oval with the “long line of the oval parallel to the lines of least skin tension” (Langer lines) (see Fig. 187-2).54 As migration of the lesion proceeds, distortion of this configuration occurs.55 The center fades after a few weeks leaving only the annular border erythematous (see eFigs. 187-2.1 and 187-2.2).
In very large lesions, only a portion of the erythematous border can be seen (see eFig. 187-2.3).
Uncommon presentations include the entire lesion being homogenously erythematous (“solidly erythematous”) (10%), forms with marked central necrosis, minimal size EM (size of the ring less than 5 cm at initial presentation) (2%), and the vesicular variant (5%).56–58
The lesion in itself is usually asymptomatic, but 50% of patients report mild tingling or itching.59 Systemic manifestations, reported in approximately 50% of patients, may appear before, during, or after the classic lesion.52,55
EM has an excellent prognosis, attributable in part to the activation of proinflammatory cytokines such as interferon-γ.60 EM usually heals spontaneously, but may persist for as long as 6–12 months; median duration in the United States is approximately 4 weeks and in Europe is approximately 10 weeks.8,19,59
Multiple EM-like lesions occur in between 1% and 17% of patients.52 The reported prevalence of multiple lesions is believed to be higher in the United States (25%–48%) than in Europe (8% or less).61 The spatial relationship of multiple lesions to the initial lesion indicates that they may be the consequence of hematogenous dissemination. Secondary EM lesions number from 2 to more than 80, usually occur away from the original lesion, and are usually smaller and less migratory (occur in “crops” of similar size, color, and shape) in comparison to classic EM.52 Lesions are usually asymptomatic and if untreated spontaneously resolve over weeks to months. Like the primary lesion, secondary EM expands over days to weeks resulting in both solid and annular erythema. Primary EM may still be present but more often a primary lesion is not found. Constitutional symptoms are usually more severe than those associated with classic EM.56
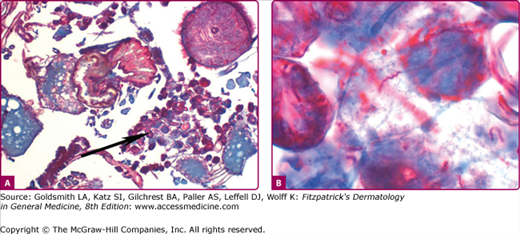
Histopathologic findings vary with the biopsy site55 and age of the lesion. Biopsies of early lesions show papillary dermal edema and a mixed infiltrate of lymphocytes, neutrophils, a few plasma cells, and a few eosinophils (Fig. 187-3). Biopsies of older lesions display a variably dense perivascular and interstitial infiltrate of lymphocytes and plasma cells (Fig. 187-4). Helpful, albeit nonspecific, clues include the presence of plasma cells and mast cells in the infiltrate.62 The presence of plasma cells and eosinophils in the same specimen is reported to be much less common than the presence of either alone.55
Figure 187-3
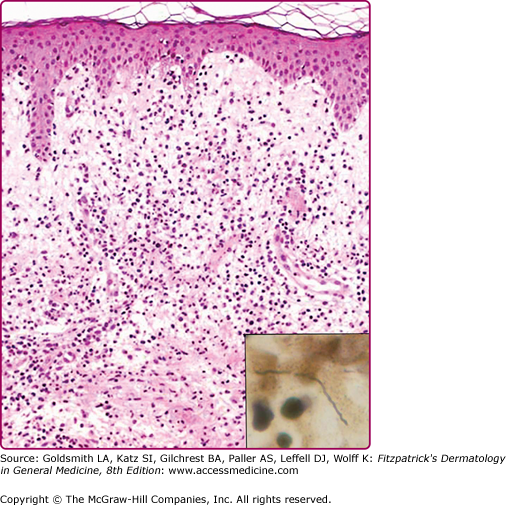
Histopathology of erythema migrans. Papillary dermal edema, mixed infiltrate of lymphocytes, neutrophils, few plasma cells, and few eosinophils (hematoxylin and eosin stain). Inset shows a spirochete in same biopsy stained with Warthin–Starry stain (oil immersion). (Reprinted with permission from Dermatopathology Interactive Atlas, edited by J Bhawan, P Sau, HR Byers, 2001.)
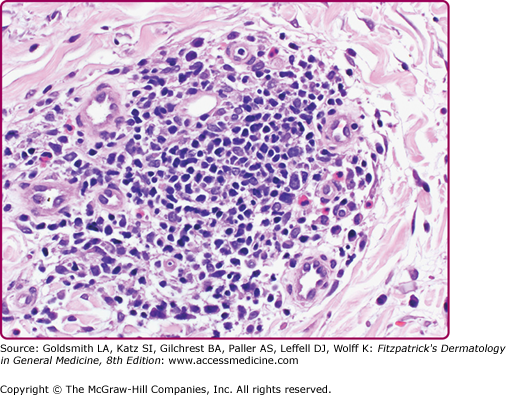
Immunohistochemical studies indicate the infiltrate to be composed of CD4+ T lymphocytes with the exception of those seen in association with HIV infection in which the infiltrate is mainly CD8+ T lymphocytes, reflective of the CD4 lymphopenia of HIV infection.63
Histopathologic features of multiple lesions of EM are identical to primary EM.
The diagnosis of EM is typically made on clinical appearance in patients in an endemic area. From biopsy specimens, spirochetes, detected using special stains, are best located in the papillary dermis and may be short or elongate at this stage of the disease (see Fig. 187-3 inset).55 Direct detection of the spirochete from biopsy specimens using newer modified immunohistochemical methods such as focus floating microscopy have a better yield in detection of organisms from EM cases. Both sensitivity and specificity are higher than for culture or polymerase chain reaction (PCR)-based techniques.64,65
Atypical lesions exist and broaden the clinical differential diagnosis to include cellulitis, tinea, other contact dermatitis, or a fixed drug reaction (Table 187-1). The differential diagnosis of vesicular EM includes poison ivy, contact dermatitis, cellulitis, herpes simplex, and impetigo.
Onset | Diagnosis/Differential Diagnosis | |
|---|---|---|
Common Manifestations | ||
Erythema migrans | Days to weeks (early) | Arthropod bite, erythema multiforme granuloma annulare, urticaria, erysipelas, brown recluse spider bite, fixed drug eruption |
Acrodermatitis chronica atrophicans | Months to years (late) | Venous insufficiency, lichen sclerosus, scleroderma, physiologic age-related atrophy, corticosteroid-induced atrophy |
Uncommon Manifestations | ||
Cutaneous scleroborrelioses | Months to years (late) | |
Morphea | Primary morphea | |
Lichen sclerosus | Primary lichen sclerosus | |
Periarticular fibrous nodules | Rheumatoid nodule, gouty tophi | |
Progressive facial hemiatrophy | Primary progressive facial hemiatrophy/Parry–Romberg syndrome | |
Eosinophilic fasciitis | Primary eosinophilic fasciitis/Shulman syndrome | |
Cutaneous atrophoborrelioses | Months to years (late) | |
Anetoderma | Primary anetoderma | |
Cutaneous lymphoborrelioses | Months to years (late) | |
B-cell dominant (including B-cell lymphoma) | Arthropod bite reaction, response to vaccination, granulomas, neoplasm | |
T-cell dominant | Pityriasis lichenoides, polymorphous light eruption | |
Other cutaneous lesions | Weeks to months to years (early or late) | |
Panniculitis | Erythema nodosum | |
Granuloma annulare | Insect bite reaction | |
Erythema multiforme | Drug eruption | |
Syphilis-like papulosquamous eruption | Secondary syphilis |
Observed mainly in elderly patients in Europe, ACA has an insidious onset and appears to have predilection for females.66 Rare cases of ACA have been reported in children.67 The time interval from the spirochete inoculation to the onset of symptoms of ACA is extremely difficult to evaluate. Most patients do not recall the specific tick bite that initiated the disease.8,68 That the spirochete can survive for decades is favored by reports indicating recovery of the spirochete from biopsies of skin from ACA patients even after 20 years.8
An inflammatory phase characterizes the early clinical stages of this biphasic disease.69 The inflammatory phase presents as a bluish-red discoloration on the extensor aspect of fingers, hands, joints, and lower extremities (see eFigs. 187-4.1 and 187-4.2).
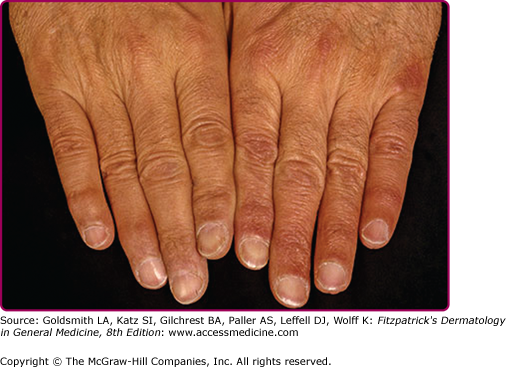
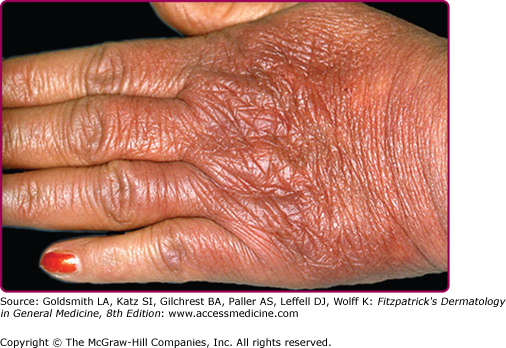
Joints commonly involved include the elbows and knees. Infiltrated purple bands of varying widths may be observed adjacent to involved joint(s). Associated findings include a cushion-like (“doughy”) swelling of the dorsum of the hands and feet (see eFig. 187-4.2).69
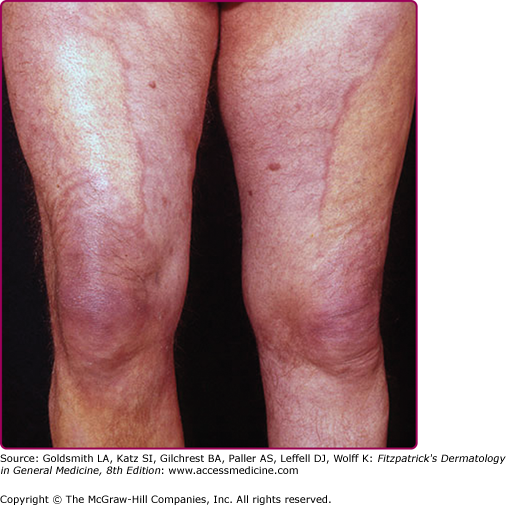
The extremities are most commonly involved, although extensive lesions on the trunk have also been documented. Lesions typically extend from the distal to the proximal portion of the extremity involved. The erythema and swelling initially vary in intensity (“waxes and wanes”), and swelling of the posterior aspect of the lower extremities is believed by some to be particularly indicative of Lyme disease.59
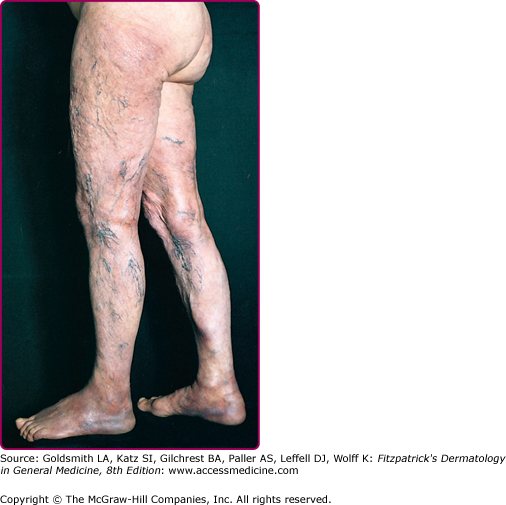
Cutaneous atrophy (Fig. 187-5), characteristic of the later clinical stage, is not an obligatory sequel to the inflammatory phase of ACA.69 Rarely, coexistence of both kinds of lesions at different sites in the same patient has been documented. The atrophic phase is characterized by lesions with a “cigarette paper-like” appearance and a prominence of superficial veins (see Fig. 187-5).
Central and peripheral nervous system involvement has also been documented in approximately 45% of patients with ACA. Thirty percent to 45% of patients suffer from a polyneuropathy, often most pronounced in the limb with cutaneous involvement.61 Chronic joint and bone involvement, attributed to persistence of spirochetes in cutaneous lesions, is most often seen in patients with long-standing ACA or an untreated lesion of EM/ACA and is typically restricted to the extremity involved. The characteristic symptom, exhibited in approximately one-third of patients in one study, was a swollen or painful foot and heel.61 Other symptoms include subluxation of small joints, bursitis, arthritis, and cortical thickening of bone. Solitary or multiple fibrotic lesions near joints, particularly in the olecranon area, may develop in some patients.69
All three species of B. burgdorferi that infect humans have been found in ACA lesions.70 Histopathologic features of biopsied lesions vary with the clinical phase of ACA. In inflammatory lesions, three layers are typically described: an atrophic epidermis, a zone of uninvolved papillary dermis, and a layer of inflammatory cells composed of lymphocytes and plasma cells.71 The presence of plasma cells in the infiltrate is documented mainly from studies from Europe, as American reports indicate that few or no plasma cells are found.68,69 The infiltrate may be deep with extension into the subcutis.59 Occasionally, interface dermatitis has been reported. Unusual findings include the presence of vacuoles, either singly or in groups, at different levels of the dermis.72 Although some believe these represent mature adipocytes, others believe them to be an expression of lymphedema, given that they are mainly observed from biopsies of markedly edematous sites. In favor of the latter hypothesis is the absence of such vacuoles from the same site posttreatment.
Phenotypic studies indicate the lymphocytes in the infiltrate are mainly of the CD4 phenotype, favoring the concept that ACA is a T cell-mediated immune response.73 Further in support of this theory is the expression of adhesion molecules, such as intracellular adhesion molecule-1, on endothelial cells, lymphocytes, and basal keratinocytes in the inflammatory infiltrate.74 Chronicity of the lesions may be partially explained by downregulation of major histocompatibility complex class II molecules on Langerhans cells.75
Detection of the spirochete using special stains, novel immunohistochemical techniques such as focus floating microscopy or PCR-based techniques substantiates the infective etiology, further supported by positive serologic findings.65 Serology is of great utility in the diagnosis of ACA. Antibodies to B. burgdorferi can be detected in almost all of patients with ACA even years after treatment.
Lesions of ACA are notoriously overlooked or misinterpreted. In those with a relevant clinical history, the onset appears to be related to an untreated lesion of EM and/or neurologic manifestations. In one study, a history of EM months to years earlier on the same side was found in approximately 18% of patients.75 Differential diagnoses are listed in Table 187-1.
Sclerotic skin lesions clinically indistinguishable from primary lichen sclerosus (LS) or morphea (see eFig. 187-5.1) develop not only in association with other dermatoborrelioses [approximately 10% of patients with ACA and borrelial lymphocytoma (see Section “Uncommon Cutaneous Manifestations of Lyme Disease”)] but also in the absence of other cutaneous manifestations of Lyme disease.56,76,77
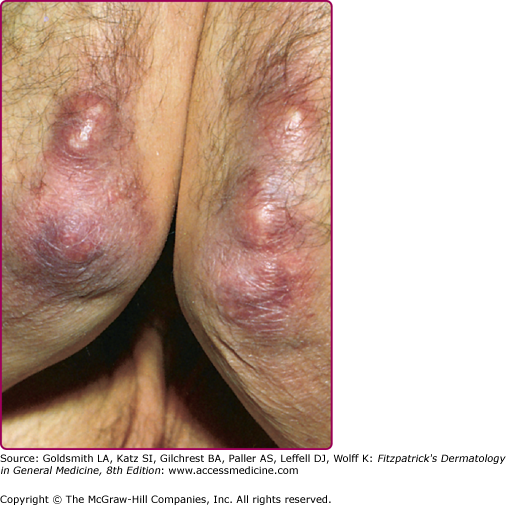
Periarticular (“ulnar”) fibrous nodules (Fig. 187-6), described in association ACA, may also occur in the absence of dermatoborrelioses.78 They usually present as hard nodules on the elbows, knees, and on the lateral aspect of the digits near joints and have been reported to be provoked by trauma, surgery, and electromagnetic radiation.79
Uncommon sclerotic disorders associated with Lyme disease include progressive facial hemiatrophy (Parry–Romberg syndrome) and eosinophilic fasciitis (Shulman syndrome) (see Chapter 157).80
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


